Эволюционная патология. К вопросу о месте ВИЧ-инфекции и ВИЧ/СПИД-пандемии среди других инфекционных, эпидемических и пандемических процессов. 2.2. Простейшие — симбионты многоклеточных
2.2. Простейшие — симбионты многоклеточных
При усложнении многоклеточных организмов сложившиеся за миллиарды лет отношения между клетками, способными к фагоцитозу и их паразитами, продолжали оставаться прежними. Этот феномен пока не изучен системно, и я вынужден для его описания использовать разрозненные экспериментальные данные разных авторов.
«Чужие здесь не ходят». Такие отношения обнаруживаются уже тогда, когда начинаешь анализировать пути проникновения в организм людей и животных микроорганизмов, вызывающих у них опасные инфекционные болезни. Не только легионеллы проникают в организм человека через макрофаги (см. подглаву 2.1 «Границы феномена сапронозного существования патогенных для людей микроорганизмов»). Эти же макрофаги «спасают» возбудитель сибирской язвы, попавший в альвеолы легких в составе мелкодисперсного аэрозоля, и переносят его в более благоприятные условия медиастинальных лимфатических узлов, где он размножается, а затем вызывает генерализованную инфекцию (Guildi-Rontani C., 2002). Возбудители брюшного тифа, дизентерии, патогенные иерсинии (Y. enterocolitica и Y. pseudotuberculosis) используют для своего выживания в организме человека посредничество М-клеток пейеровых бляшек (Agmina s. insulae Peyeri) подвздошной кишки. Дальнейшая их диссеминация происходит исключительно благодаря выживанию внутри макрофагов, которые мигрируют через лимфатическую систему (Smith H., 1996; Finlay B., Falkow S., 1997). Возбудитель туляремии проникает в макрофаги при любом механизме инфицирования и с их «помощью» в течение нескольких дней поражает все ретикулоэндотелиальные ткани (Ellis J., Green M., 2002). Возбудитель бруцеллеза инфицирует как фагоцитарные клетки — макрофаги, так и нефагоцитарные — трофобласты беременных животных (Ko J., Splitter G. A., 2003). Сам организм теплокровного хозяина является для этих микроорганизмов биологическим тупиком.
Возбудитель сибирской язвы. В настоящее время экология Bacillus anthracis изучена плохо и обычно ее описание ограничивается упоминанием того, что микроорганизм может долго существовать в почве и хорошо растет на мясопептонных средах (см. например, работы Шуваловой Е. П. с соавт., 2001; Таршиса М. Г. и Черкасского Б. Л., 1997). Но из-за его высоких поражающих свойств как агента биологического оружия, патогенез сибирской язвы у людей и животных изучен очень обстоятельно (см. работы: Dixon T. D., et al., 1999; Prince A. S., 2003; Guarner J. et al., 2003).
Эндоспоры Bacillus anthracis могут проникать в макроорганизм любыми путями (чрезкожно, аэрогенно, энтерально и др.), но все их пути ведут в макрофаги, выступающие как универсальные входные ворота инфекции (рис. 28).
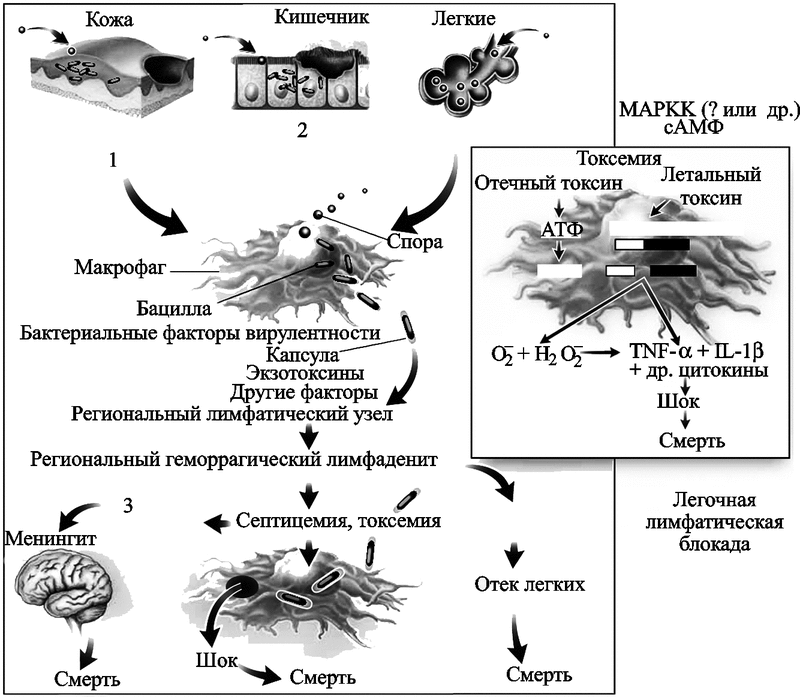
Рис. 28. Патогенез сибиреязвенной инфекции у млекопитающих
Макрофаги поглощают бациллы и мигрируют к региональным лимфатическим узлам. Вегетативные B. anthracis растут в лимфатических узлах, вызывая регионарный геморрагический лимфаденит. Бактерии распространяются через кровь и лимфу по всему организму и вызывают большое число смертельных исходов из-за септицемии. В случаях легочной формы болезни, перибронхиальные геморрагические лимфадениты блокируют легочный лимфатический дренаж, вызывая отек легких. Смерть наступает от септицемии, токсемии или легочной недостаточности и может произойти через 1-7 суток после заражения.
1 - низкоуровневое прорастание и рост в участке инфицирования ведут к локальному отеку и некротическому поражению кожи;
2 - низкоуровневое прорастание и рост в участке инфицирования ведут к массивному выпоту, отеку слизистой и некротическому поражению кишечника;
3 - лимфагенное и гематогенное распространение B. anthracis.
MAPKK (mitogen-activated protein kinase kinase) - митоген-активированный белок киназы киназы; TNF - фактор некроза опухолей; IL - интерлейкин (Dixon T. D., et al., 1999).
Отношения B. anthracis с эволюционными предшественниками макрофагов, свободноживущими почвенными амебами, в процессе эволюции сложились как паразитические, т. е. они их использовали для своего размножения, разрушали и сохранялись в окружающей среде в виде спор. После заглатывания спор другими простейшими, цикл повторялся. Такой вывод можно сделать, не имея пока данных о взаимоотношении сибиреязвенного микроба с простейшими почв исходя из наблюдений за его поведением в альвеолярных макрофагах млекопитающих. Макрофаги поглощают споры B. anthracis посредством фагоцитоза (рис. 29.).
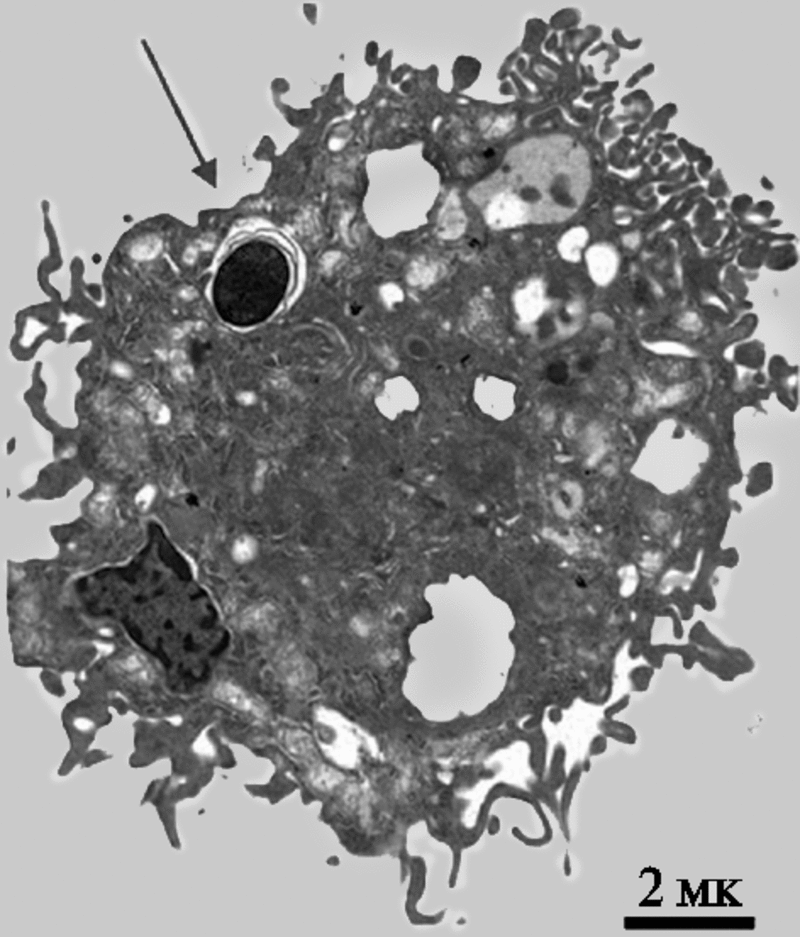
Рис. 29. Фагоцитоз спор B. anthracis альвеолярными макрофагами (AM)
AM были инфицированы спорами B. anthracis (штамм Sterne) и через 30 мин исследованы с помощью трансмиссионного электронного микроскопа. Положение споры внутри AM показано стрелкой.
Черная полоса соответствует 2 микронам (Ribot W. et al., 2006).
Инфицировав альвеолярные макрофаги, споры быстро достигают эндосомальных компартментов, где должно происходить их разрушение. Однако некоторые споры остаются жизнеспособными и после прорастания превращаются в бациллы, которые реплицируются внутри макрофагов. По данным S. Ruthel et al. (2004), это происходит следующим образом. Временные интервалы «прорастания» спор варьировали, но обычно бациллы начинали удлиняться через 4—5 часов после проникновения в альвеолярные макрофаги, и реплицировались уже через 5—6 часов. В их экспериментах до 10 % макрофагов оказались неспособными подавить репликацию сибиреязвенного микроба. Среднее количество спор на макрофаг, которое в последующем приводило к репликации бактерий, составило 6—7, следовательно, более высокая множественность заражения повышала возможность преодоления спорами защитной способности макрофагов.
Форма макрофагов была типичным отражением роста бацилл внутри макрофагов: плазматическая мембрана макрофагов растягивалась, чтобы вместить все бациллы, и примерно через 7 часов макрофаг разрывался. Споры образовывали цепочки еще внутри клеток, до их гибели. В организме животных этот процесс завершался уже в лимфатических узлах (Ruthel S. et al., 2004). Следовательно, альвеолярные макрофаги не запускают каскад иммунных реакций, направленных на освобождение организма от возбудителя сибирской язвы, а «пособничают» ему, доставляя его в более безопасные для размножения ткани, но так им было предопределено естественным отбором еще в архее.
В начале 1990-х гг. установлено, что и токсины B. anthracis действуют на организм человека опосредованно, через макрофаги. Когда у мышей удаляли макрофаги, они приобретали устойчивость к сибиреязвенному токсину (Hanna P. C. et al., 1993). Вегетативные B. anthracis секретируют два экзотоксина. Отечный токсин является кальмодулин-зависимой аденилат-циклазой (calmodulin-dependent adenylate cyclase), увеличивающей внутриклеточные уровни циклического AMP (cAMP) при проникновении в большинство типов клеток. В месте введения токсина в ткани экспериментальных животных нарушается водный гомеостаз и появляется массивный отек. Летальный токсин (LT) представляет собой цинкметаллопротеазу (zinc metalloprotease), которая вызывает гипервоспалительное состояние макрофагов, активируя пути окислительного взрыва и высвобождая активированные кислородом интермедиаты, а также продукцию провоспалительных цитокинов, таких как фактор некроза опухолей альфа (TNF-альфа) и интерлейкин-1бета, ответственные за шок и смерть больного (Dixon T. D. et al., 1999).
Размножению B. anthracis в альвеолярных макрофагах способствует летальный токсин. Подвергнутые его воздействию макрофаги не утрачивают способности поглощать споры возбудителя сибирской язвы, однако в значительной мере теряют способность освобождаться от них (Ribot W. et al., 2006).
На первый взгляд, продуцирование B. anthracis токсинов свидетельствует лишь об одном зловещем предназначении этого микроорганизма — массовые убийства людей и животных на территориях, с древности называемых «проклятыми полями», другое с антропоцентристских представлений об эпидемических процессах нелогично. Но еще Ю. В. Вертиев (1996) обратил внимание на сходство структуры и механизма действия бактериальных токсинов, интерферонов, бактериоцинов и гормонов. Эти вещества синтезируются одним типом клеток, в то время как воздействуют на другие типы клеток в чрезвычайно низкой концентрации (10-11-10-14 М). Они обладают сходной молекулярной организацией — состоят как минимум из двух функционально и структурно различных белков: энзиматического и рецепторного; имеют сходные звенья молекулярного механизма действия (связывание с рецепторами, активация, транслокация внутрь клетки и модификация клеточных мишеней); обладают сходной кинетикой биологического эффекта — одноударный эффект; и, наконец, все эти вещества в определенных дозах токсичны.
Если предположить, что способность бактерий синтезировать токсины закрепляло какую-то неизвестную сегодня сигнальную функцию в образуемом ими биоценозе, то понятен и двухкомпонентный состав, и одноударность их действия (по бинарным токсинам бацилл см. обзорную работу Barth H. et al., 2004). Преимущество такой структуры для передачи сигналов заключается в том, что при ее распространении из центра сигнал не ослабляется на большом расстоянии. Если бы передача сигнала осуществлялась структурами, не способными к лиганд-специфическому взаимодействию, то сигнал ослабевал бы по мере диффузии сигнальных молекул. Отсюда, как следствие, способность воздействовать на другие типы клеток в чрезвычайно низких концентрациях. Применительно к почвенным свободноживущим амебам такое действие должно заключаться в блокировании каких-то их защитных функций, облегчающих микроорганизму проникновение в эти организмы и последующее размножение. Выброс почвенными фагоцитирующими клетками хемокинов — это сигнал о помощи, адресованный другим простейшим, своего рода призыв двигаться к месту нападения «на своих». У многоклеточных организмов это свойство хемокинов используется для направленной миграции лейкоцитов в очаг воспаления (см. подглаву 2.2 «Реликтовая иммунная система человека»).
Возбудители бруцеллеза. Виды Brucella (B. melitensis, B. suis, abortus, B. canis) являются опасными патогенами сельскохозяйственных животных и человека и рассматриватся учеными в качестве потенциальных агентов биологического оружия. Об экологии бруцелл известно еще меньше, чем об экологии возбудителя сибирской язвы. Считается, что бруцеллез является исключительно зоонозной инфекцией. Имеются экспериментальные данные, свидетельствующие о значительной устойчивости бруцелл во внешней среде (Шувалова Е. П. с соавт., 2001; Таршис М. Г., Черкасский Б. Л., 1997).
О взаимоотношениях бруцелл с простейшими, естественно, ничего неизвестно, однако их взаимоотношения с макрофагами изучены весьма обстоятельно и, что весьма важно для достижения цели нашей работы, они между ними складываются совсем иначе, чем у B. anthracis.
Бруцеллы обитают в организме млекопитащих преимущественно внутри макрофагов и плацентарных трофобластов. Хронический характер вызываемых Brucella инфекций обусловлен в основном их способностью к длительному существованию в фагосомном компартменте фагоцитирующих клеток их хозяев (рис. 30).
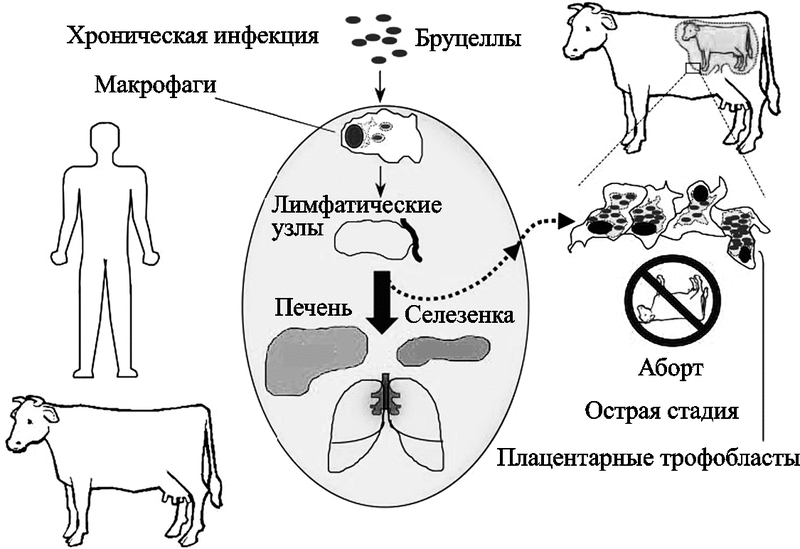
Рис. 30. Основные типы клеток, являющиеся местом обитания B. abortis у млекопитающих (Roop R. M. et al., 2004)
R. M. Roop et al. (2004) называют их способность выживать и размножаться в макрофагах «потрясающей» («impressive»). Их удивляет то обстоятельство, что, хотя опсонизация специфическими IgG или активация гамма-интерфероном повышает бруцеллацидную активность культивированных макрофагов, вирулентные штаммы все же выживают в этих клетках и демонстрируют внутриклеточную репликацию. Механизм выживания бруцелл в макрофагах следующий.
Сразу после проникновения в макрофаг бруцеллы располагаются в подкисленном компартменте, который сливается с компонентами ранней стадии эндосомного обмена. Там они переживают окислительный взрыв. Поэтому нет ничего удивительного в том, что в экспериментах они весьма устойчивы к действию факторов внешней среды, хотя с ней сами непосредственно не соприкасаются. Продвижение содержащих бруцеллы вакуолей по эндосомно-лизосомному пути обмена ограничено, и вирулентные штаммы переносятся во внутриклеточный компартмент, обычно называемый репликативной фагосомой. Иногда его еще называют «бруцеллосомой». Репликативные фагосомы возникают в результате обычного клеточного процесса — постоянных взаимодействий между содержащей Brucella вакуолью и эндоплазматическим ретикулумом макрофагов хозяина. Эти репликативные фагосомы не только не сливаются с лизосомами, но и внутри их повышается рH; условия среды становятся более благоприятными для внутриклеточного существования бруцелл (рис. 31).
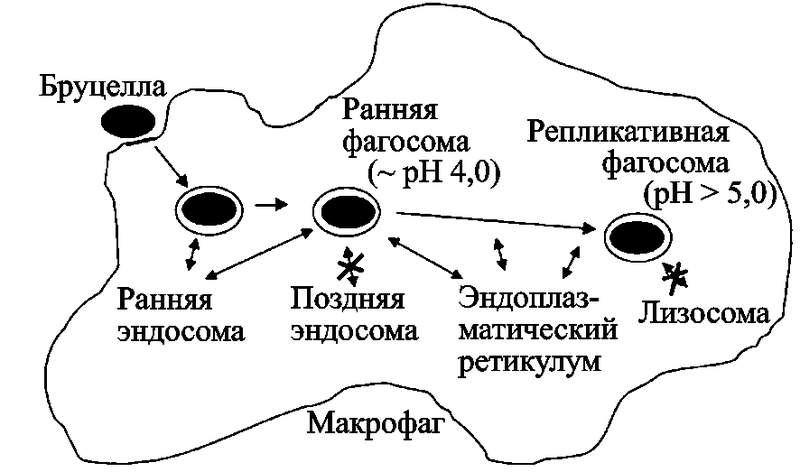
Рис. 31. Биогенез репликативной фагосомы, являющейся местом обитания B. abortus в культивированных мышиных макрофагах (Roop R.M. et al., 2004)
Недавно было продемонстрировано, что гладкий липополисахарид (ЛПС) В. abortus обладает иммуномодулирующей активностью, которая потенциально помогает бактерии в обеспечении длительного существования в макрофагах хозяина. В частности, эта молекула не разрушается фагоцитами, а переносится к их клеточной поверхности, где она образует макродомены с молекулами МНС класса II. Образование комплекса «МНС класса II—ЛПС» приводит к снижению способности инфицированного хозяина активировать специфичные к антигенам Brucella CD4+ Т-клетки (Forestier et al., 2000).
Отсутствие этапа слияния с лизосомами сближает репликативный цикл бруцелл с репликативным циклом легионелл (см. рис. 22), и делает его непохожим на репликативный цикл возбудителя сибирской язвы. Бруцеллы имеют совершенно иной набор генов «факторов вирулентности», чем B. anthracis. Большинство из таких идентифицированных генов необходимы не для нанесения микроорганизмом «поражения» фагоцитирующей клетке, а для физиологической адаптации к условиям среды в ее репликативной фагосоме. Например, это гены, кодирующие фермент цитохром-bd-оксидазу, имеющий высокий аффинитет к кислороду и позволяющий бруцеллам выживать в условиях окислительного стресса. Оперон bvrRS Brucella кодирует двухкомпонентную регуляторную систему, контролирующую экспрессию генов, содействующих сохранению целостности клеточной оболочки. Измененные свойства мембраны bvrRS-мутантов В. abortus делают их менее устойчивыми к кислой среде, в которой им приходится находиться в фагосомном компартменте макрофагов хозяина на ранних стадиях инфекции. Обнаружено также, что bvrRS-мутанты В.abortus неспособны достичь репликативной фагосомы (более подробно о таких «факторах вирулентности» см. в работе Roop R. M. et al., 2004).
Для нас в цикле размножения бруцелл в макрофагах важно другое — проглядывающееся многообразие отношений между прокариотами и эукариотическими одноклеточными почвенными обитателями. В отличие от B. anthracis, бруцеллы ведут эндосимбиотический образ жизни в макрофагах и, соответственно, в свободноживущих почвенных амебах, что неизбежно сказывается на клинике вызываемой ими болезни. Бруцеллез — хроническое и длительное страдание, сопровождающееся изнурительной лихорадкой. В настоящее время не существует безопасной и эффективной вакцины, которую можно использовать для предупреждения развития бруцеллеза у людей. Лечение этой болезни антибиотиками требует длительного применения их определенной комбинации. К тому же число случаев возврата болезни после завершения курса антибиотикотерапии достигает 10% — сложившиеся в архее эндосимбиотические отношения бруцелл с фагоцитирующими клетками пока выше возможностей человека.
Возбудитель туляремии. Экспериментально установлена способность возбудителя этой болезни, F. tularensis, поддерживаться в почвенных простейших A. castellanii (см. рис. 25) и макрофагах (рис. 32).
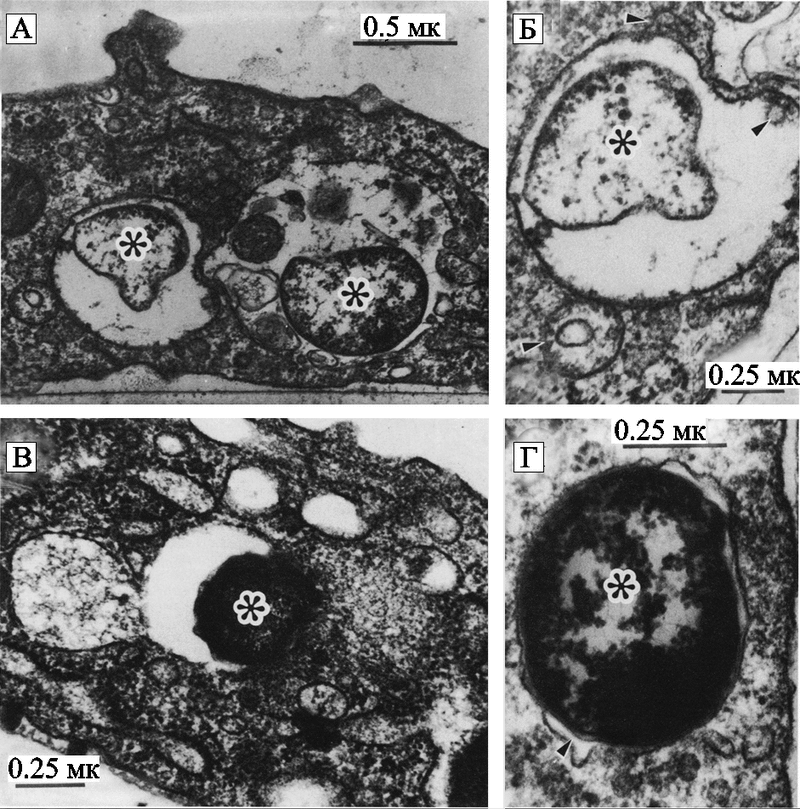
Рис. 32. Трансмиссионное электронно-микроскопическое исследование макрофагов, инфицированных Francisella sp.
(A) F. novicida в пределах фагоцитирующей везикулы. Черная полоса соответствует 0.5 мк.
(B) Детали фагоцитирующей везикулы, содержащей F. noivicida в пределах макрофага. Характерны небольшие везикулы в пределах крупной везикулы и небольшие светлые везикулы, ассоциированные с наружной поверхностью везикулы (стрелки). Черная полоса соответствует 0.25 мк.
(C) Фагоцитирующая везикула макрофага, содержащая F. tularensis LVS. Черная полоса соответствует 0.25 мк.
(D) Детали фагоцитирующей везикулы, содержащей F. tularensis LVS. Характерным является то, что периплазматическое пространство и наружную мембрану можно различить, так как мембрана ограничена фагоцитирующей везикулой. Черная полоса соответствует 0.25 мк. Везикулы содержат бесформенный или электронно-яркий материал, обычно ассоциированный с содержащими бактерии везикулами. Микроорганизмы обозначены звездочкой (Antony L. et al., 1991).
L. Antony et al. (1991) изучили рост в макрофагах грызунов F. tularensis и близкородственного микроорганизма F. novicida. Ими обнаружено, что хотя представители Francisella sp., попав в макрофаги, выживают в пределах фагосом по одинаковому механизму, не сливаясь с лизосомами (также как бруцеллы и легионеллы), но макрофаги разных видов грызунов они «различают». Так, возбудитель туляремии оказался способным расти в макрофагах мышей, крыс и морских свинок; F. novicida размножалась в макрофагах мышей и морских свинок, но не крыс. Этот феномен косвенно свидетельствует о существовании у представителей таксона Francisella разных хозяев среди одноклеточных в природных резервуарах.
Показательно и то, что F. tularensis убивает своих случайных хозяев-макрофагов точно таким же образом, как и паразиты свободноживущих амеб — размножившись, они индуцируют апоптическую гибель клетки-хозяина. В отношении макрофагов так поступают многие патогенные для человека микроорганизмы — Salmonella, Yersinia, Shigella, Legionella, Brucella. Правда, у каждого из них свой сценарий апоптоза, зависящий от их жизненного цикла в фагоцитирующих клетках (Xin-He Lai et al., 2001; Fernandez-Prada C. V., 2003).
Возбудитель чумы. До обнаружения ВИЧ, по своей способности вызывать катастрофические эпидемии он не знал равных. Относится к потенциальным агентам биологического оружия и уже неудачно использовался японской армией в военных целях в годы Второй мировой войны. С конца XIX столетия считается, что возбудитель чумы поддерживается в природе среди грызунов. У отдельных ученых подозрения о сапронозном существовании Yersinia pestis появились гораздо раньше, еще тогда, когда не только не существовало этого термина, но и сам возбудитель чумы не был открыт. Возможность поддержания возбудителя чумы среди простейших показана в 1991 г. (см. подглаву 2.1 «Простейшие и их паразиты» и нашу книгу: Супотницкий М. В., Супотницкая Н. С., 2006).
Экспериментальные доказательства того, что Y. pestis проникает в организм человека, затем выживает, размножается и распространяется по органам и тканям с помощью макрофагов, получены почти 50 лет назад (Cavanaugh D. C., Randall R., 1959). Везикулы, содержащие Y. pestis, обычно обнаруживают в фаголизосомах (рис. 33).
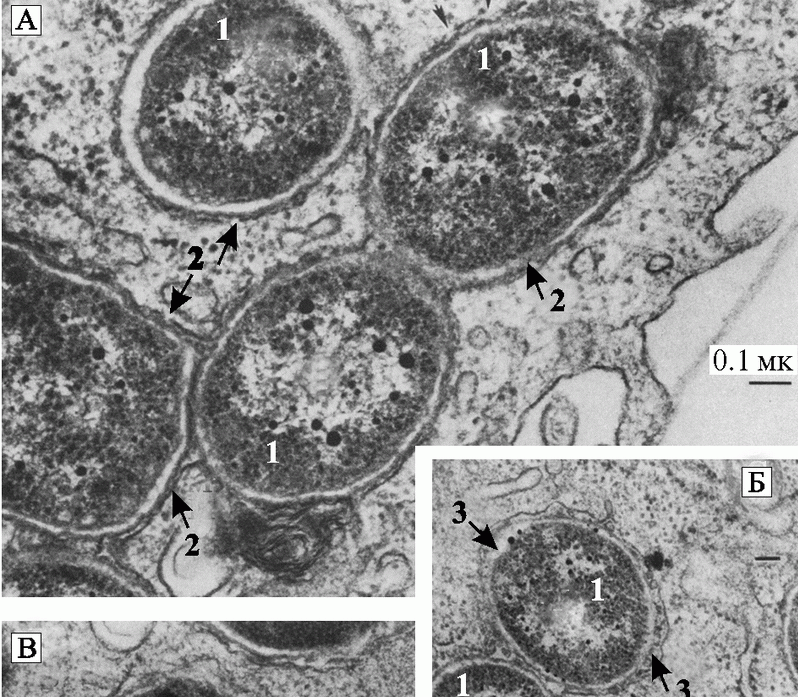
Рис. 33. Трансмиссионная микрофотограмма Y. pestis в фаголизосомах перитонеальных макрофагов мышей
Снимки сделаны на поздней стадии инфекции. Вокруг внутриклеточно расположенных Y. pestis (Y) всегда находится мембрана (М). На изображениях В и С показаны крошечные везикулы (V), часто ассоциирующиеся с бактериями внутри мембраны. Черная полоса соответствует 0,1 мк (Straley S., Harmon P., 1984).
Показано, что продолжительность существования Y. pestis в пределах макрофагов во время инфекционного процесса зависит от природы хозяина и инфицированных тканей. До 80 % фагосом перитонеальных макрофагов, населенных Y. pestis, сливаются с лизосомами в течение ближайших 2,5 ч после инфицирования, процесс заканчивается в течение 8 ч. Но за это время возбудитель чумы формирует вместительную вакуоль и успевает размножиться (Grabenstein J. et al., 2006). Следовательно, в патогенезе чумной инфекции проявляется эволюционно закрепленная за Y. pestis способность к паразитическому существованию среди почвенных простейших, когда микроорганизм не вступает в эндосимбиотические отношения со своим хозяином. Но мир простейших очень сложен и разнообразен. Возможно, что среди одних простейших возбудителю чумы удается существовать как эндосимбионту, среди других, ставших эволюционными предшественниками макрофагов, он размножается как паразит. Экспериментально это пока никак не подтверждено, но такие альтернативы в существовании Y . pestis могут объяснить как некоторые особенности чумных эпидемий, так и клиники самой болезни.
Для эпидемий чумы, воспринимаемых современниками как катастрофические (пандемия «черной смерти» в 1346-1351 гг., вспышки чумы в Лондоне в 1665 г., в Марселе в 1721 г., в Москве в 1771 г. и др.), характерны:
1) постепенное распространение болезни по территории;
и 2) цикличность эпидемического процесса.
Первые случаи болезни вызываются маловирулентными возбудителями, сотни людей ходят с бубонами, но смертность среди них невысока. Затем вирулентность Y. pestis повышается, смертность среди людей нарастает, бубоны «затвердевают», болезнь проявляет себя в крайне тяжелых формах, по восприятию современников чума на этой стадии эпидемического цикла не поддается лечению, но, достигнув определенного пика, эпидемия резко идет на убыль. Болезнь вновь проявляется в относительно легкой форме. Врачи получают возможность сообщать о своих успехах в лечении чумы с помощью порошка из крабьего глаза, «уксуса четырех разбойников» или просто выжигая бубон раскаленной кочергой.
Первую особенность эпидемий чумы можно объяснить проникновением Y. pestis из экосистем «простейшие-Y. pestis», в которых она существует как эндосимбионт; в простейшие, где она размножается как паразит. Если количество таких простейших в среде, окружающей экосистему «простейшие-Y. pestis», по каким-то причинам достигло неизвестной нам критической массы, то Y. pestis попадает в окружающую среду (в почву). Корнями трубчатых растений бактерия «выносится» на поверхность во все возрастающих количествах. Стебли этих растений поедают грызуны и инфицируются Y. pestis. Далее через эктопаразитов инфицировавшихся грызунов возбудитель чумы проникает в дома людей и вызывает среди них вспышку чумы.
Вторая особенность эпидемий чумы заключается в постепенном нарастании вирулентности Y. pestis (см. нашу книгу: Супотницкий М. В., Супотницкая Н. С., 2006). По аналогии с объяснением повышения вирулентности возбудителя холеры среди гидробионтов, данным Пушкаревой В. И. и Литвиным В. Ю. (см. подглаву 2.1 «Границы феномена сапронозного существования патогенных для людей микроорганизмов»), этот феномен можно объяснить клональной селекцией вирулентных штаммов Y. pestis среди простейших, в которых она ведет паразитическое существование. Снижение вирулентности Y. pestis вызвано как повышением резистентности самих свободноживущих простейших, так и грызунов, вовлеченных в процесс передачи этого микроорганизма людям (гибель высокочувствительных к Y. pestis грызунов и простейших, формирование иммунных к возбудителю чумы популяций грызунов и др.). Высоковирулентные штаммы возбудителя чумы сами обрывают цепочки, по которым они распространяются из активизировавшихся реликтовых очагов.
Клиника болезни у человека или грызуна, предопределяется паразитированием Y. pestis среди макрофагов. Возбудитель из места проникновения (например, места укуса чумной блохи) заносится в ближайшие лимфатические узлы, они воспаляются. Из кровеносной системы в инфицированный лимфатический узел рекрутируются другие макрофаги, так формируется бубон, заполненный останками погибших макрофагов. Размножившиеся Y. pestis поступают из лимфоузлов в кровь (септицемия); выброс разрушающимися макрофагами огромного количества лимфокинов, нехарактерного для иммунных реакций, протекающих в варианте нормы и к которым адаптирован организм, дает картину шока и помрачения сознания больных. У людей отдельных генотипов развиваются кожные (карбункулы, петехии, геморрагии) и/или легочные поражения (вторичная легочная чума). Так как процесс разрушения макрофагов протекает быстро и возбудитель чумы поступает в кровь, где он доступен действию антибиотиков, то своевременно начатая антибиотикотерапия обычно дает эффект (в отличие от антибиотикотерапии при бруцеллезе, возбудитель которого вступает в эндосимбиотические отношения с макрофагами; см. выше). Поэтому сегодня смертность при бубонной чуме составляет не более 8 %, вместо 50-80 % в эпоху «до антибиотиков».
В лимфоидных тканях при чумной инфекции происходят процессы, которые берут начало еще во времена господства на Земле одноклеточных организмов. В весьма содержательных экспериментах Marketon M. et al. (2005) было обнаружено, что в селезенке мыши при экспериментальной чумной инфекции практически не поражаются Т- и В-клетки, хотя они там представляют большинство. Зато «в полном составе» гибнут такие клетки-мишени, как макрофаги, дендритные клетки и гранулоциты/нейтрофилы. Авторы делают вывод, что Y. pestis, используя систему секреции III типа для разрушения клеток с врожденными иммунными функциями, добивается ускорения летального исхода при чуме. Невольно возникает вопрос к этим авторам, а что, Т- и В-клетки у нас не обладают иммунными функциями, раз возбудитель чумы их «не замечает»? Ведь именно Т-клетки при взаимодействии с макрофагами инициируют у них эффективное слияние фагосом, захвативших бактерии, с лизосомами, разрушающими внутриклеточные патогены и выступают «организаторами» комплексного иммунного ответа на возбудитель чумы. И именно антигенраспознающие рецепторы В-клеток запускают синтез антител, специфичных к структурам Y. pestis. К феномену отбора возбудителями опасных болезней клеток иммунной системы в качестве мишеней мы еще вернемся в главе 3.
Вирус натуральной оспы (ВНО). ВНО тысячелетиями был спутником человека, он встречал его после рождения, и нередко сразу же провожал в «последний путь», что сегодня напоминает «выбраковку» Природой отдельных особей вида Homo sapiens. Вызываемая ВНО болезнь оказалась единственной в истории эпидемий, которую с древности считали «очистительной» (см. работу Губерта В., 1896). А так как натуральная оспа до введения массовой противооспенной вакцинации поражала почти всех детей до года, то ее до начала XVIII столетия не относили к инфекционным болезням, а считали «врожденным» заболеванием, которое обязательно должно проявится у ребенка после рождения. Например, Авиценна (см. его труд «Канон врачебной науки», кн. 4) утверждал, что оспа есть результат брожения в крови человека, сходное с брожениями, которые происходят в выжатых соках плодов и приводят к тому, что их частицы отделяются одна от другой. Причиной такого борожения крови ребенка являются «остатки питательного вещества месячных женщины, образовавшегося при беременности или зародившегося после нее от мутных, нехороших яств, которые разжижают состав крови и волнуют ее».
В настоящее время натуральная оспа считается уничтоженной. Мы не нашли данных, свидетельствующих о возможности поддержания ВНО среди почвенных амеб, но его поведение среди макрофагов ничем не отличается от поведения тех микроорганизмов, для которых такая связь установлена, впрочем, судите сами.
ВНО среди людей распространяется воздушно-капельным путем. В организме человека он предварительно накапливается в альвеолярных макрофагах, затем по лимфатическим путям проникает в лимфатические узлы, где происходит его репликация, после чего ВНО обнаруживается в свободных макрофагах иммунной системы и в крови (первичная виремия). Дальнейшее его распространение происходит по лимфатической системе, и, только накопившись в фагоцитах, он в больших количествах попадает в кровоток (вторичная виремия). Даже если ВНО попадает в организм человека нехарактерным для него путем, например, через порез в коже, его накопление осуществляется фагоцитирующими клетками. Сначала он проникает в макрофаги, ими заносится в лимфатические узлы, там размножается и затем распространяется по организму (Маренникова С.С., Щелкунов С.Н., 1998) (более подробно о генерализации ВНО см. подглаве 3.1).
Микобактерии. Ежегодно в мире выявляется до 8 млн новых случаев туберкулеза среди людей (Hui Pan et al., 2005) и от 500 до 700 тыс. новых заболевших «малозаразной» болезнью — проказой (Scollard D. M. et al., 2006). Сегодня имеются веские основания считать возбудители туберкулеза (M. tuberculosis) и проказы (M. leprae) сапронозами (см. подраздел 2.1). Эти два микроорганизма, несмотря на свое таксономическое сходство, вызывают у людей разные болезни, а их распространение в человеческих популяциях проявляется разными эпидемическими процессами. Ниже мы рассмотрим элементы их прошлого сапронозного существования в инфекционном процессе. В подразделе 4.1 обратимся к механизмам, которые вовлекают эти микроорганизмы в эпидемии.
M. tuberculosis. Ежегодно в мире погибает около 2 млн больных туберкулезом (Hui Pan et al., 2005). Возбудитель болезни проникает в организм человека через альвеолы, куда он попадает в составе капелек аэрозоля. Далее возбудитель туберкулеза диссеминируется по организму посредством фагоцитирующих клеток. Прежде всего, это альвеолярные макрофаги, клетки альвеолярного эпителия II типа — пневмоциты (pneumocytes), они присутствуют в альвеолах в гораздо большем количестве, чем макрофаги, и могут попадать в желудочно-кишечный тракт человека; и дендритные клетки (Smith I., 2003). Последние в фундаментальных руководствах обычно описываются с точки зрения осуществления ими «иммунного надзора» в нелимфоидных органах и тканях, где они захватывают антигены и процессируют их в пептиды. После этого, процессированные антигены связываются с антигенпредставляющими молекулами дендритных клеток (белки классов I и II главного комплекса гистосовместимости), те представляют эти антигены цитотоксическим лимфоцитам и Т-хелперам, и «иммунный надзор» пресекает развитие инфекционного процесса (см., например, руководство М. А. Пальцева, 2004).
Возможно, эта схема работает против каких-то других микроорганизмов, но только не в отношении «старых знакомых» фагоцитирующих клеток — M. tuberculosis и M. leprae. В отличие от дифференцированных макрофагов, дендритные клетки обладают способностью к активной миграции по тканям организма и поэтому их считают важным фактором в развитии инфекционного процесса при туберкулезе и проказе (Smith I., 2003; Scollard D. M. et al., 2006).
Однако вернемся к M. tuberculosis. В легочных тканях инфицированные фагоцитирующие клетки в пределах от 2 до 6 недель вызывают клеточные иммунные ответы, проявляющиеся «наплывом» лимфоцитов и активацией и повреждением макрофагов, формирующих так называемую гранулему — казеозные очаги, окруженные фибриновой тканью. Казеозную («творожистую»; caseum = cheese) массу гранулемы формируют погибшие макрофаги (более подробно о формировании и классификации таких гранулем см. в работе Grosset J., 2003). Жизнеспособные M. tuberculosis обычно сохраняются в казеозном центре гранулемы и могут реактивироваться и инфицировать дыхательные пути, вызывая некроз бронхов и образуя полости в легочной ткани. Фиброзные изменения легочной ткани соответствуют отчаянной попытке защитных механизмов хозяина остановить инфекционный процесс путем формирования вокруг центральной зоны некроза защитного вала из коллагена, эластина и гексозаминов (Schluger N. W., 2005).
Роль альвеолярных макрофагов в этом процессе следующая. M. tuberculosis связывается с ними посредством рецепторов комплемента (CR1, CR2, CR3 и CR4), маннозных рецепторов и других поверхностных молекул, хорошо «знакомых» микобактерии из их совместного эволюционного прошлого (например, toll-like receptor 2; TLR2) (Schluger N. W., 2004). Взаимодействие бациллы с маннозным рецептором фагоцитирующей клетки происходит через поверхностный микобактериальный гликопротеин липоарабиноманн (lipoarabinomannan, LAM) (Schlesinger L. S., 1994). M. tuberculosis проникает в макрофаг посредством эндоцитирующей вакуоли — фагосомы (Smith I., 2003).
Туберкулезная палочка не имеет классических факторов вирулентности, подобных токсинам Corynebacterium diphtheriae, Escherichia coli O157:H7, Shigella dysenteriae или Vibrio cholerae, определяющих клинику болезни. Однако микобактерия ингибирует фагосомно-лизосомное слияние [phagosome–lysosome (P-L) fusion] еще в момент своего контакта с фагоцитирующей клеткой. P. Kang et al. (2005) установили, что взаимодействие липоарабиноманна микобактерии с маннозным рецептором является ключевым моментом предотвращения фагосомно-лизосомного слияния. Благодаря ему предотвращается формирование кислой среды во внутриэндосомальном пространстве и блокируется увеличение концентрации Ca2+ в цитозоле макрофага. Так как увеличение концентрации Ca2+ стимулирует другие защитные реакции клетки-хозяина на инфекцию, например, такие как «респираторный взрыв», продукция NO и цитокинов, то блокирование этой реакции помогает M. tuberculosis избежать срабатывания защитных механизмов хозяина не только на уровне макрофага, но иммунной системы в целом. Такая «выносливость» туберкулезной бациллы в ранней эндосоме уменьшает экспрессию белка главного комплекса гистосовместимости II типа на поверхности макрофага и презентацию антигенов M. tuberculosis Т-хелперам, что приводит к длительному персистированию этого микроорганизма среди клеток реликтовой иммунной системы (объяснение термина «клетки реликтовой иммунной системы» приведено в подглаве 2.3). Детально механизм, посредством которого патогенные микобактерии предотвращают созревание фагосомы, не изучен (Smith I., 2003). Однако микобактерии могут существовать в фагоцитирующих клетках в больших количества и это никак не отражается на прогрессировании болезни (Kaushal D. et al., 2002). Неконтролируемый рост M. tuberculosis, начинающийся обычно из сайта инфекции, приводит к обширным разрушениям легочной ткани и гибели пациента из-за кислородной недостаточности и легочного кровотечения. Неблагоприятный исход болезни может быть связан с облитерацией легочной паренхимы из-за хронической кислородной недостаточности, с обструкцией бронхо-альвеолярной проходимости в результате гранулематозного роста и по другим причинами (Шебанов Ф.В., 1964). Их мы рассмотрим в подразделе 4.1.
M. leprae — возбудитель «малозаразной» инфекционной болезни — проказы (другое название — лепра). «Малозаразной» она считается с позиций контагионистических представлений, предполагающих, что источником возбудителя болезни является больной человек, но эпидемические цепочки между случаями заражения людей удается проследить далеко не всегда. Общее количество таких больных в мире в 1988 г. приближалось к 11 млн, из них 62% приходилось на Азию, 34 % на Африку (Hastings R. еt al., 1988). В начале этого столетия, по данным ВОЗ, их насчитывалось уже 15 млн человек (Шувалова Е.П. с соавт., 2001). Сами механизмы заражения человека M. leprae далеко не так очевидны, как казалось еще несколько лет тому назад. Например, A. Hagge et al. (2004) и D. M. Scollard et al. (2006) вообще предпочли не давать их конкретного объяснения, указав на то, что механизмы трансмиссии возбудителя проказы между людьми неизвестны. Неизвестны так же инфицирующая доза возбудителя болезни, точное количество инфицированных M. leprae людей, так как до появления первых клинических признаков болезни нет возможноcти установить сам факт их инфицирования; а соответственно нет полной ясности и по границам эндемичных по проказе регионов мира.
Таксономическое сходство M. leprae с M. tuberculosis свидетельствует об удивительном многообразии взаимоотношений в мире микроорганизмов. Геном M. leprae не превышает 3.3 Mb (миллионов оснований), тогда как у M. tuberculosis он достигает 4.4 Mb. У возбудителя проказы снижено соотношение содержания G+C (58 % у M. leprae, против 66 % у M. tuberculosis). И главное, M. leprae содержит только 1614 открытых рамок считывания генов и 1133 псевдогенов, в отличие от 3993 функционирующих генов и 6 псевдогенов, имеющихся у M. tuberculosis. Массив функционирующих генов M. leprae почти на 50 % меньший, чем у M. tuberculosis, у которой около 90 % генома представлены функционирующими генами. Такое упрощение генома M. leprae проявляется элиминированием целых метаболических путей. У возбудителя лепры вырождена защита от токсических радикалов, отдельные гены, определяющие механизмы функционирования детоксикации, являются псевдогенами, ни один из дополнительных 142 генов, найденных только у M. leprae, не связан с метаболическими путями. M. leprae не способна поглощать железо из окружающей среды (см. http://www.leprosy-ila.org/Mycobacterium.html). Ее до сих пор не научились культивировать на искусственных питательных средах.
Дегенеративность генома M. leprae сказывается на скорости ее размножения в клетках человека. Ее удвоение в макрофаге требует от 10 до 20 суток. У M. tuberculosis этот процесс занимает не более 20 ч, E. coli в жидкой искусственной питательной среде делится за 20 мин (Bjune G. et al., 1983). Сравнение геномов возбудителей туберкулеза и проказы показывает более глубокую специализацию M. leprae к пока еще неизвестному природному резервуару среди простейших, чем это можно ожидать для M. tuberculosis.
Более 95 % людей устойчивы к проказе. M. leprae проникает в организм человека через повреждения кожных покровов или поверхность слизистых оболочек. В большинстве случаев внедрившиеся бациллы погибают и не вызывают болезни у человека. Однако в отдельных случаях, которые мы детально рассмотрим в подразделе 4.1, развивается латентная лепра, которая может переходить в ту или иную клиническую и иммуногистопатологическую форму болезни (лепроматозная, туберкулоидная и недифференцированная) в зависимости от характера иммунной реакции организма человека на возбудитель болезни. Лепроматозная лепра (lepromatous leprosy, LL) развивается тогда, когда иммунный ответ проявляется Th2-типом цитокинового профиля с плохо выраженным клеточным иммунитетом. На противоположном конце спектра иммунных ответов находится туберкулоидная лепра (tuberculoid leprosy, TT) с выраженным клеточным иммунным ответом и Th1-типом цитокинового профиля. Между этими двумя полярными формами лепры располагаются различные смешанные варианты ее течения (Hagge A. et al., 2004).
Для специфической гранулемы лепроматозного типа характерно наличие крупных «лепрозных клеток» (клеток Вирхова) с вакуолизированной протоплазмой и массой заключенных в них M. leprae. Кроме того, в состав гранулемы входят лимфоциты, плазматические клетки, фибробласты и изредка тучные и гигантские клетки. Между инфильтратом и нижней границей эпидермиса обычно остается узкая полоска неизмененной соединительной ткани. В период обострения болезни (лепроматозная реакция) присоединяются сосудистые изменения, гнойно-фибриноидная экссудация, затем расплавление с участием эозинофилов и лейкоцитов. Гранулема туберкулоидной проказы типична, но не специфична. Она состоит из эпителиоидных клеток с примесью гигантских и окружена лимфоцитарным валом. Около сильно измененных нервных веточек встречаются единичные бациллы M. leprae. В фазе обострения (туберкулоидная реакция) кровеносные сосуды расширены, наблюдается интерстициальный отек, в центре гранулемы — фибиноидный некроз и скопление лимфоцитов. Поражения недифференцированного типа состоят из лимфоцитов и небольшого количества гистиоцитов и фибробластов. Инфильтрат располагается преимущественно периневрально. Нервные клетки подвергаются восходящим дегенеративным и деструктивным изменениям. Бациллы M. leprae обнаруживаются не всегда, и их, как правило, мало (Торсуев Н. А., 1964).
Как и M. tuberculosis, M. leprae паразитирует в фагоцитирующих клетках и способна подавлять их взаимодействие с Т-хелперами (подробнее об иммунных реакциях на возбудитель проказы см. в работе D. M. Scollard et al., 2006). В основном это дендритные клетки, макрофаги и Шванновские клетки нервной ткани. M. leprae распространяется по тканям преимущественно дендритными клетками. Макрофаги являются основным хозяином M. leprae в организме человека. С поверхностью макрофагов человека возбудитель проказы связывается посредством рецепторов тех же типов, что и M. tuberculosis: маннозных (C-type lectin receptors, CD206), Toll-подобных и рецепторов комплемента (CR1 и CR3 на поверхности моноцитов; и CR1, CR3, и CR4 на поверхности макрофагов). Также им блокируется слияние фагосомы с лизосомой, «респираторный взрыв», продукция NO и цитокинов, что позволяет M. leprae избежать быстрой гибели в фагосоме (Scollard D. M. et al., 2006).
Макрофаг не убивает M. leprae, и если Т-специфический иммунитет активируется, как это имеет место у больных с лепроматозной формой болезни, макрофаги остаются неактивированными, и размножение бацилл в них продолжается (см. выше). Затем макрофаг разрывается, и M. leprae инфицирует новые макрофаги, «собравшиеся» в участок поражения (Hagge A. et al., 2004). В одном макрофаге можно насчитать 100 и более лепрозных бацилл, но это не сказывается на его жизнеспособности. В условиях in vitro при температуре 330 С макрофаг может поддерживать M. leprae неделями (Scollard D. M. et al., 2006). Это предпочтительная для возбудителя проказы температура тканей человека, поэтому лепрозные поражения в основном располагаются в холодных участках поверхности кожи (Sasaki S. et al., 2001). Лепроматозный участок кожи может содержать до 1010 бацилл M. leprae на грамм ткани (Hastings R. et al., 1988).
Шванновские клетки (леммоциты) представляют собой клетки нервной ткани, образующие оболочки длинных отростков нервных клеток (аксонов) в периферических нервах и ганглиях. Филогенетически это очень древняя ткань — подобная оболочка окружает периферические нервы моллюсков и червей. Шванновские клетки похожи на простейшие организмы, они имеют реснички и способны к волнообразным движениям. В отношении отростков нервных клеток они выполняют опорную функцию. Через вещество шванновских клеток или на их стыке в отросток нервной клетки проникают метаболиты.
M. leprae проникает в шванновскую клетку путем взаимодействия с G-доменом альфа2-цепи ламинина (laminin 2) и вызывает поражения нервной системы на поздней стадии болезни (Rambukkana A. et al., 1998). Взаимодействие происходит за счет двух адгезинов клеточной стенки микобактерии — фенольного гликолипида (phenolic glycolipid-1, PGL-1) и LBP21, гистоноподобного протеина, закодированного у M. leprae геном ML1683 (Barker L., 2006). А дальше уже сама M. leprae начинает наращивать свою экологическую нишу, как это она делает в своем первичном природном резервуаре. Установлено, что путем индукции циклина D1 (сyclin D1) и p21, двух ключевых регуляторов G1-фазы клеточного цикла, внутриклеточная M. leprae вызывает размножение немиелинизированных (non-myelinated) Шванновских клеток (Tapinos N., Rambukkana A., 2005) — механизм, который никак не может сформироваться в результате коэволюции M. leprae и Homo sapiens.
Ретровирусы. ВИЧ-инфекция проявляется очевидной патологией — нарушением функционирования иммунной системы. Поэтому с самого начала изучения этой проблемы интерес у ученых вызывали только те феномены, которые имеют отношение к взаимодействию вируса с клетками иммунной системы человека и их последствиям. Такая позиция понятна, если воспринимать события, происходящие с участием иммунной системы человека, во-первых, в его, человека, ощущении времени; во-вторых, если считать наши представления об иммунной системе законченными, нуждающимися лишь в мелких дополнения. А чтобы эта позиция стала еще прочнее, надо просто не учитывать то обстоятельство, что ВИЧ в принципе не может научиться управлять иммунной системой человека, так как у них обоих нет возможности для совместной эволюции и «учебы». ВИЧ, если он вызывает инфекционный процесс у хозяина, то уже не оставляет после себя живых и чему-то «научившихся» (коэволюционировавших) особей.
Роль тканевых макрофагов в патогенезе ВИЧ-инфекции также оказалась очень не похожей на ту, которую им положено играть в иммунном ответе на возбудитель инфекционной болезни, придерживаясь учебников по иммунологии. Было установлено, что они не только не «переваривают» ВИЧ, но и «перетаскивают» его в лимфатические узлы и ткани мозга (Koening S. et al., 1986; Kaul M. et al., 2005; Trujillo R. et al., 2007) (рис. 34).
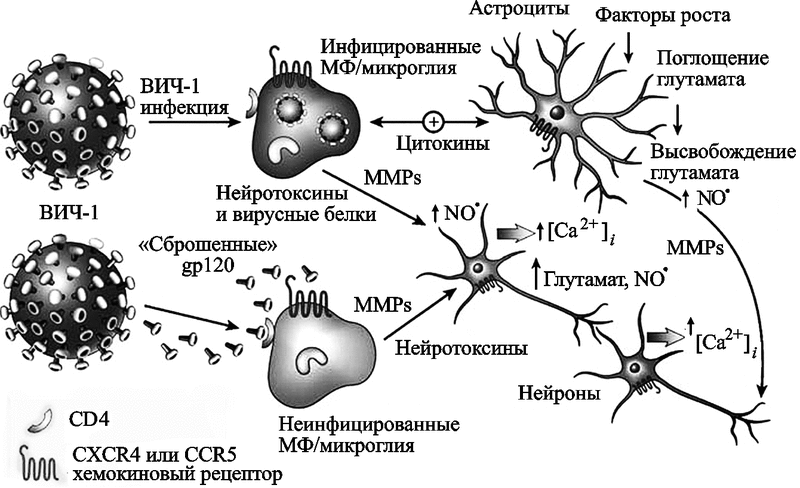
Рис. 34. Современная модель повреждения и смерти нейронов под воздействием ВИЧ-1-инфекции
ВИЧ проникает в ЦНС с макрофагами. Клетки макрофагальных линий составляют большинство из ВИЧ-1-инфицированных клеток мозга, которые формируют многоядерные гигантские клетки, гистопатологический признак продуктивной ВИЧ-1 инфекции в мозге. Инфицированные макрофаги (MF)/микроглия высвобождают различные нейротоксические субстанции. MMPs (matrix metalloproteinases) - матриксметаллопротеазы.
Более подробно о механизмах повреждения мозга при ВИЧ-инфекции см. в работе Kaul M. et al., 2005.
Еще макрофаги играют резервуарную роль для ВИЧ, делая его недоступным ни для средств антиретровирусной терапии, ни для специфических антител (Orenstein J. M. et al., 1997). Но этим «странности» во взаимоотношениях ВИЧ с фагоцитирующими клетками человека не кончаются, они плавно переходят в пробелы в представлениях об эволюции нашего вида (см. подглаву 2.3).
Выше на конкретных примерах показана способность фагоцитирующих клеток человека и почвенных простейших взаимодействовать с микроорганизмами и друг с другом посредством одних и тех же рецепторов — маннозных ( mannose receptor, MR ) (см. подглаву 2.1 «Простейшие и их паразиты»). Но и ВИЧ демонстрирует этот же путь проникновения в фагоцитирующие клетки человека. После проникновения ВИЧ в макрофаги посредством маннозного рецептора, его дальнейшая судьба напоминает судьбу представителей Brucella sp . — он становится внутриклеточным эндосимбионтом. Такая возможность у ВИЧ существует благодаря его поверхностному гликопротеину gp 120, лишь наполовину представляющему собой белковую молекулу. Оставшуюся половину составляют гликозилированные карбонгидратные группы. Функциональное значение этих остатков сахаров было продемонстрировано через исследование исключительной плотности гликозилирования оболочки HIV-1, предотвращающей связывание и нейтрализацию вируса антителами, но не связывание с маннозными рецепторами макрофагов ( Wei X. et al ., 2003).
R. Trujillo et al. (2007) установили, что избирательность ВИЧ-1 по отношению к специфическим типам клеток является регулируемой посредством взаимодействия между вирусной оболочкой и клеточными рецепторами. Когда ВИЧ проникает в клетку посредством CD4 и других хемокиновых рецепторов, специфически взаимодействующих с gp120 оболочки вируса, то его «поведение» в клетке становится другим — он начинает реплицироваться, активно используя ресурсы клетки, т. е. «превращается» в паразита (рис. 35).
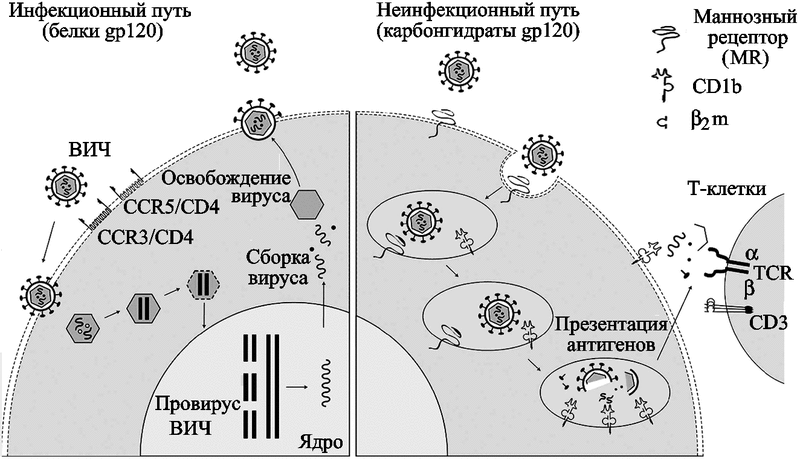
Рис. 35. Модели проникновения ВИЧ-1 в макрофаги/микроглию
Неинфекционный путь HIV-1 — в присутствии MR (правая схема), богатые маннозой карбонгидраты gp120 инициируют проникновение вируса в макрофаг. Этот путь проявляет себя презентацией антигенов вируса через CD1b-антиген, и индуцирует клеточный иммунитет.
Инфекционный путь HIV-1 (левая схема) регулируется через протеин/протеиновое взаимодействие между gp120 ВИЧ-1 и клеточными белковыми рецепторами, сгруппированными с корецепторами.
MR - маннозный рецептор (Trujillo R. et al., 2007).
Аналогично Plasmodium (P. knowlesi и P. vivax) ВИЧ использует рецепторы хемокинов, известные как DARC (рецептор хемокина, связанный с антигеном Даффи). DARC экспрессируется эритроцитами, капиллярными эндотелиальными клетками, эпителиальными клетками собирательных почечных трубочек, легочными альвеолами и мозжечковыми клетками Пуркинье (Pogo A. O., Chaudhuri A., 2000), что позволяет эритроцитам действовать как ретровирусный резервуар (Lachgar A. et al., 1998). Любопытно то, что этот рецептор очень древний, так как он взаимодействует с хемокинами C-X-C и C-C подклассов (Lachgar A. et al., 1998), чья эволюционная история начинается в архее (см. подглаву 2.3).
ВИЧ имеет собственный белок, вызывающий хемотаксическую активность моноцитов человека — Nef. Ранее он был известен как фактор, способствующий прогрессированию СПИДа. Но M. H. Lehmann et al. (2006) уточнили механизм его действия. Они установили, что внеклеточный Nef дозозависимо увеличивает миграцию моноцитов; благодаря этому механизму ВИЧ распространяется по организму человека.
Ретровирусы в лабораторных условиях легко инфицируют модельных животных через дыхательные пути. В естественных условиях такой путь передачи не только им не нужен, но и противоречит их стратегии паразитизма (см. подглаву 3.3). Эти результаты можно объяснить по аналогии с вышеприведенными данными следующим образом. ВИЧ легко проникает в альвеолярные макрофаги модельных животных потому, что он уже хорошо знаком с их эволюционными предшественниками — свободноживущими амебами почв.
В пользу последнего предположения также свидетельствуют:
1) многократное инфицирование приматов на протяжении их эволюционной истории ретровирусами одного семействе (см. подглаву 1.3);
2) наличие в геноме отдельных «молодых» видов млекопитающих «древних» ретровирусов приматов.
Например, получены доказательства, по крайней мере, двух «взрывов» распространения ERV-L по геному мышей, но не крыс, имевших место менее чем 10 млн лет назад. Однако в геноме приматов ERV-L «обосновались» не менее чем 45 млн лет назад (Benit L. et al., 1999). Естественно, что мыши не могут быть эволюционной ветвью приматов и инфицировались они ERV-L уже после дивергенции их предкового вида на крыс и мышей. Поэтому объяснить наличие в их геноме копий этих ретровирусов от какого-то общего предка грызунов, а не от источника «вне вида», поддерживающего эти ретровирусы в экзогенном состоянии миллионы лет, весьма затруднительно.
***
Ни один из перечисленных в подглаве микроорганизмов, включая ВИЧ, не нуждается для своего поддержания в природе в существовании человека как биологического вида. Поэтому не может быть и речи об их совместной эволюции. Даже те механизмы их взаимодействия, которые производят впечатление «хорошо пригнанных» друг к другу, «пригонялись» в тот период геологического времени, когда в природе господствовали простейшие-эукариоты и прокариотические организмы. ВИЧ взаимодействует с фагоцитирующими клетками человека с использованием тех же механизмов, что и другие паразиты свободноживущих простейших. В то же время поддержание экзогенных ретровирусов клетками реликтовой иммунной системы многоклеточных организмов проявляется другим процессом, эволюционным. Мы рассмотрим его ниже.