Препараты, полученные из крови человека и животных, в аспекте показателей качества, эффективности и безопасности

(в соавторстве с А.А. Елаповым, И.В. Борисевичем, Э.Ю. Кудашевой, В.И. Климовым, Е.В. Лебединской, Л.В. Корсун, Е.В. Горбуновой, В.Г. Слободяном, В.А. Меркуловым, Ю.В. Олефиром)
Проблемы качества и безопасности препаратов, получаемых из плазмы крови человека и гипериммунной сыворотки животных, решаются строгой государственной регламентацией процессов их производства. Проблема безопасности таких препаратов сводится к минимуму путем их очистки от примесей, вызывающих осложнения у пациентов (агрегаты иммуноглобулинов, протеазы, плазмин, плазминоген, активатор прекалликреина, примеси IgA и IgM и др.). К настоящему времени разработаны способы фракционирования минорных белков крови, использующие их многоэтапное разделение аффинной хроматографией, исключающие этап осаждения этанолом при низких значения рН (осаждение по Кону). Вирусная безопасность достигается многоступенчатым процессом производства, включающим не менее двух независимых методов очистки крови от вирусов (сольвент-детергентная обработка и ультрафильтрация) и самого продукта путем аффинной хроматографии. Обосновано предположение, что к настоящему времени достигнут предел развития базовых технологий получения препаратов из плазмы крови человека и из гипериммунных сывороток животных. Их дальнейшее развитие не предполагает выхода за пределы частных усовершенствований технологий получения и очистки препаратов крови (препараты для подкожного применения, комбинации различных иммуноглобулинов в препарате, повышения эффективности выделения минорных белков и др.) и повышения их вирусной и прионовой безопасности. В тоже время нерешенность этих проблем и потребности рынка предполагают появление принципиально новых препаратов, полученных генно-инженерным путем, хорошо охарактеризованных по молекулярному составу, обладающих высокой избирательность в отношении мишеней воздействия. Это рекомбинантные факторы крови с измененными свойствами; коктейли из рекомбинантных антител и Fab-фрагментов IgG, высокоафинных к эпитопам токсинов и др. Поэтому в ближайшие годы в России необходимо создавать принципиально новую систему оценки качества, эффективности и безопасности препаратов крови, учитывающую дальнейшее направление их развития.
Библиографическое описание: Супотницкий МВ, Елапов АА, Борисевич ИВ, Кудашева ЭЮ, Климов ВИ, Лебединская ЕВ, Корсун ЛВ, Горбунова ЕВ, Слободян ВГ, Меркулов ВА, Олефир ЮВ. Препараты, полученные из крови человека и животных, в аспекте показателей качества, эффективности и безопасности. Биопрепараты 2015; (3): 33–48.
The problems of quality and safety products derived from human blood plasma and hyperimmune animal sera as well as recombinant blood products resolved strict government regulation of their production processes. The risk of implications is minimized by plasma fractionation and purification of a specific drugs from various impurities (immunoglobulin aggregates, protease, plasmin, plasminogen, prekallikrein activator, IgA and IgM etc.). Viral safety is achieved by multi-step manufacturing process that includes at least two independent methods (treatment with solvent/detergent + incubation at low pH or pasteurization, combined with polyethylene glycol processing). It was justified that for today the technological process of the development of plasma preparations and hyperimmune animal sera has reached its limit. Their further development is the most likely to refer to specific improvements. The improvements will relate to increasing the efficiency of manufacturing technologies and methods of clinical use (preparations for subcutaneous administration, combinations of different immunoglobulin preparations, etc.), viral safety, ways to eliminate component, that were previously not considered to be able to influence the outcome of clinical use (soluble molecules CD4, CD8, HLA, thrombin, trace amounts of blood clotting factors VIII, IX, X, XI, XII etc.). At the same time new genetic engineered preparations with well-characterized molecular composition and a high selectivity for target impact are expected to appear on the market because of these unsolved issues. These are recombinant blood factors with altered properties; cocktails of recombinant antibodies and Fab-fragments of IgG, highly affine for toxin epitopes, etc. Therefore, in the upcoming years it is necessary to create in Russia a new system for assessing the quality, efficacy and safety of blood products, taking into account the future course of their development.
Bibliographic description: Supotnitskiy MV, Elapov AA, Borisevich IV, Kudasheva EYu, Klimov VI, LebedinskayaEV, Korsun LV, Gorbunova EV, Slobodyan VG, Merkulov VA, Olefir YuV. Blood preparations of humans and animals in terms of their quality, efficacy and safety. Biopreparation (Biopharmaceuticals) 2015; (3): 33–48.
Препараты, полученные из крови человека и животных, используются для лечения инфекционных болезней, поражений токсинами, различных форм гемофилии, иммунологических и метаболических расстройств, врожденной или приобретенной гипопротеинемии и компенсации потерь крови. К сожалению, до конца 1980-х гг. их применение в клинике приводило к многочисленным передачам вирусных инфекций и тяжелым осложнениям неинфекционного характера. Производителям не хватало знаний, позволяющих провести научную оценку возможной связи показателей качества, эффективности и безопасности препаратов крови с используемыми технологическими процессами. В настоящее время ситуация с безопасностью и качеством таких препаратов изменилась в лучшую сторону, однако возрос масштаб их производства и ассортимент выпускаемой продукции, само производство интернационализировалось, что предполагает значительно большие последствия от случайной ошибки, чем это было 30 лет назад. Цель данной работы – анализ перспективности подходов к получению препаратов из плазмы крови в аспекте показателей качества, эффективности и безопасности.
Поиск информации проводился на глубину 20 лет. Использовалась информационно-поисковая система PubMed. В качестве источников информации отбирались обзорные и прогнозные статьи, подготовленные ведущими специалистами в данной области. Для упрощения работы по выявлению тенденций развития технологий получения препаратов из плазмы крови, прежде всего, анализировалась научная литература по так называемым «опережающим объектам», т.е. объектам, исследование которых вследствие их большей общественной потребности, продвинулось дальше, чем по другим объектам данной группы [1, 2]. В качестве таких объектов по способам получения препаратов из плазмы крови человека рассматривались иммуноглобулины для внутривенного введения (ВВИГ); по способам получения гетерологичных иммуноглобулинов – противозмеиные, противоботулинические, антидифтерийные и антистолбнячные иммуноглобулины; по рекомбинантным препаратам крови – рекомбинантные факторы VIII и IX.
1. Иммуноглобулины человека
Иммуноглобулины человека представляют собой лекарственные средства, содержащие антитела против возбудителей инфекционных болезней или их токсинов.
Иммуноглобулины для внутривенного введения. Наиболее часто используемые препараты, изготовленные из плазмы крови доноров. Государственный реестр лекарственных средств и Анатомо-терапевтическо-химическая (Anatomical Therapeutic Chemical) классификация (АТХ) относят ВВИГ к фармацевтической группе «медицинские иммунобиологические препараты» (код по АТХ JO6BA02) [3].
Современные ВВИГ получают фракционированием плазмы крови человека. Они представляют собой препараты поликлональных антител класса IgG, синтезированных В-лимфоцитами в ответ на антигенные стимулы, имевшие место на протяжении жизни человека-донора. IgG – гликопротеин с молекулярной массой около 150 кДа, содержащийся в плазме человека в количестве от 7 до 12 г⁄л [4]. Класс IgG классифицируют на четыре подкласса (IgG1, IgG2, IgG3, IgG4), класс IgA – на два подкласса (IgA1, IgA2). Все классы и подклассы составляют девять изотипов, которые присутствуют в норме у всех индивидов. Каждый изотип IgG определяется последовательностью аминокислот константной области тяжелой цепи [5].
Современные препараты ВВИГ подразделяются на три группы [3, 6]:
I. Стандартные препараты – содержат в основном IgG (иммуноглобулин человека нормальный для внутривенного введения).
II. Стандартные специфические (гипериммунные) препараты – содержат в основном IgG, но имеют более высокое содержание противовирусных антител.
III. Обогащенные препараты ВВИГ – содержат антитела классов IgG, IgM, IgA против патогенных вирусов и бактерий.
Терапевтический эффект от введения пациенту стандартного препарата ВВИГ не сводится только последствиям возмещения до физиологической нормы IgG в сыворотке крови. Эффективность и безопасность его медицинского применения определяются дуализмом функции IgG: они могут специфически взаимодействовать с чужеродными антигенами; и одновременно способны вызывать неспецифические эффекты. Такая функциональная дихотомия является следствием особенностей структуры молекулы IgG. Ее вариабельный регион (два Fab-фрагмента) состоит из легкой и частично из тяжелой цепей и специфически взаимодействует с антигенами, что обусловлено меняющейся от белка к белку последовательностью аминокислотных остатков в N-концевой части молекулы. Константный регион (Fc- или кристаллизующийся фрагмент) связывает компонент комплемента 1 (С1) и взаимодействует с Fc-рецепторами макрофагов или нейтрофилов. Активация эффекторной функции Fc-фрагмента антитела происходит после агрегации IgG на поверхности антигена, структура молекулы меняется, что служит сигналом для запуска системы комплемента или индукции опсонизации через фагоцитоз (рисунок 1) [5–7].
Механизмы терапевтического действия интактного IgG изучены фрагментарно. Marcia et al. [8] выделяют следующие направления воздействия ВВИГ на иммунную систему человека, сопровождающиеся терапевтическим эффектом (рисунок 2):
взаимодействие с Fc-участком специфического рецептора (FcR);
контроль запуска системы комплемента и активация механизмов растворения иммунных комплексов, циркулирующих в кровяном русле;
формирование идиотип-анти-идиотип димеров (idiotype-anti-idiotype dimer);
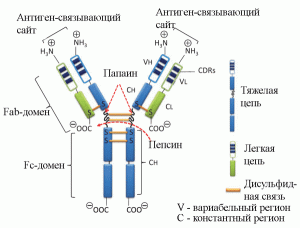 Рисунок 1. Схематическое строение иммуноглобулина G. Обработка пепсином приводит к расщеплению в участке молекулы на С-концевой стороне, за дисульфидной связью, соединяющей две тяжелые цепи вариабельного участка IgG. В результате образуются один сдвоенный F(аb')-фрагмент [F(аb')2] и один Fc-фрагмент. Расщепление IgG папаином происходит в N-концевом участке, непосредственно перед дисульфидной связью, в результате образуются два одинаковых Fab-фрагмента и один Fc-фрагмент [4]
Рисунок 1. Схематическое строение иммуноглобулина G. Обработка пепсином приводит к расщеплению в участке молекулы на С-концевой стороне, за дисульфидной связью, соединяющей две тяжелые цепи вариабельного участка IgG. В результате образуются один сдвоенный F(аb')-фрагмент [F(аb')2] и один Fc-фрагмент. Расщепление IgG папаином происходит в N-концевом участке, непосредственно перед дисульфидной связью, в результате образуются два одинаковых Fab-фрагмента и один Fc-фрагмент [4]

модулирование синтеза отдельных цитокинов и их антагонистов;
апоптоз B- и T-клеток через активацию Fas-рецептора;
блокирование взаимодействия между Т-клетками и суперантигенами;
ингибирование дифференциации и «созревания» дендритных клеток;
стимулирвание регуляторных Т-клеток (regulatory T cells, Tregs);
ингибирование дифференциации и амплификации TH17-клеток, приводящее к снижению ими продукции воспалительных цитокинов и других медиаторов воспаления.
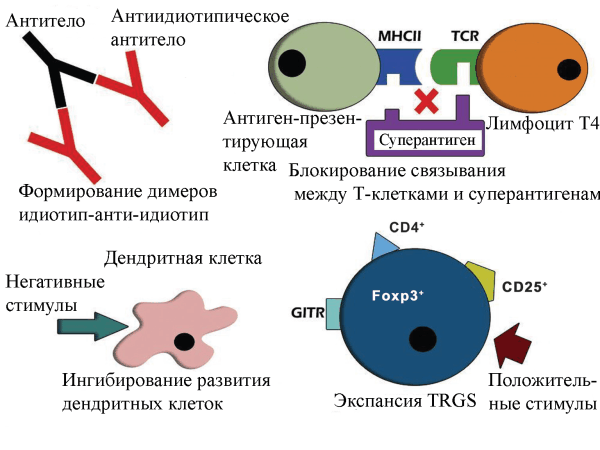
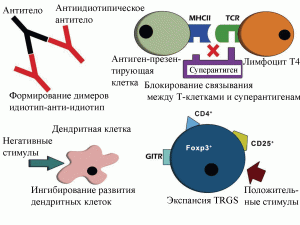 Рисунок 2. Механизмы терапевтического действия ВВИГ [8]. А – формирование идиотип-анти-идиотип димеров; Б – блокирование взаимодействия между Т-клетками и суперантигенами; В – ингибирование дифференциации и «созревания» дендритных клеток; Г – стимулирование регуляторных Т-клеток.
Рисунок 2. Механизмы терапевтического действия ВВИГ [8]. А – формирование идиотип-анти-идиотип димеров; Б – блокирование взаимодействия между Т-клетками и суперантигенами; В – ингибирование дифференциации и «созревания» дендритных клеток; Г – стимулирование регуляторных Т-клеток.
Обобщение опыта клинического применении ВВИГ позволило Донюш [3] утверждать, что они обеспечивают:
- увеличение бактерицидной активности сыворотки, стимуляцию фагоцитоза, нейтрализацию некоторых бактериальных токсинов;
- блокаду дифференцировки В лимфоцитов, продуцирующих антитела;
- предотвращение или блокаду взаимодействия аллергена с IgE, фиксированного на тучной клетке, за счет IgG4 блокирующих антител;
- подавление продукции аллерген-специфических и ауто-антител за счет воздействия антиидиотипических антител;
- снижение продукции и активности провоспалительных цитокинов;
- предупреждение комплемент-зависимого повреждения тканей за счет связывания C3b и C4b компонентов комплемента;
- предохранение от дополнительных вирусных инфекций, обладающих триггерным эффектом при аутоиммунных заболеваниях.
Первыми неспецифическими иммуноглобулинами, использованными в клинической практике, были иммуноглобулины для внутримышечного применения (intramuscular immunoglobulin, IMIG). В России разрешены иммуноглобулин человека нормальный, противоаллергический и 6 специфических иммуноглобулинов, получаемых из плазмы крови иммунизированных людей (противооспенный, антирабический, антистафилококковый, противостолбнячный, против гепатита В и клещевого энцефалита) [6].
Технология приготовления таких иммуноглобулинов разработана в 1940-х гг. Она включает этапы получения плазмы крови человека, отделения криопреципитата (содержит в основном липиды, липопротеины, углеводы, ДНК и пигменты) и осаждения IgG этанолом при температуре ниже 0 оС и определенном значении рН [5–7]. Дополнительной очистки IgG не проводилось. Получаемый препарат содержал 70–80% мономерных IgG и значительные количества IgA и IgM. Вводимые в его составе в организм человека антитела имели обычный период полураспада, активировали комплемент в присутствии антигена и обладали опсонизирующими свойствами. Применение нормальных иммуноглобулинов оказалось эффективным для профилактики и лечения кори, гепатита А и для предупреждения бактериальных инфекций у детей с наследственной агаммаглобулинемией [3, 4].
Непреодолимыми в рамках данной технологии получения IgG недостатками данных препаратов, стали болезненность в месте введения, низкая скорость поступления антител в системный кровоток и невозможность быстро создавать высокие концентрации антител в ургентных ситуациях. При попытках внутривенного введения у пациентов развивались опасные анафилактоидные реакции и гипотония, что связано с неспецифической активацией комплемента в результате спонтанного образования агрегатов иммуноглобулинов и наличием в препарате следовых количеств протеаз [8]. Поэтому применение препаратов, полученных по данной технологии, ограничено внутримышечным введением.
Попытки повысить безопасность ВВИГ в 1960-х гг. привели к значительному снижению их терапевтической эффективности. Сделав правильный вывод о ключевой роли Fc-фрагмента IgG в неспецифической активации комплемента, разработчики предприняли попытки получить ВВИГ либо без такого фрагмента, либо инактивировав его в составе IgG. Для этого IgG расщепляли пепсином, но F(аb')2 исчезали из кровотока в течение 2 суток; расщепление плазмином приводило к получению моновалентных F(аb') с низкой нейтрализующей способностью; не удавалось добиться и полной инактивации Fc-фрагмента в составе IgG обработкой алкилирующими и ацетилирующими соединениями. К тому же энзиматические и химические модификации IgG приводили к утрате ими важной физиологической функции – активации комплемента комплексом «антиген-антитело», что необходимо для эффективного лизиса бактерий и вирусов, поглощенных лейкоцитами [4]. В настоящее время выпуск таких препаратов прекращен [5].
Низкая терапевтическая эффективность энзиматических и химических производных IgG вынудила разработчиков ВВИГ в начале 1970-х гг. вернуться к получению интактного IgG. Проблемы качества и безопасности ВВИГ на основе интактного IgG решались строгой государственной регламентацией процессов сбора и фракционирования плазмы доноров, контроля производства. Были разработаны национальные и международные документы, регулирующие производство ВВИГ. Система таких мер приведена в таблице 1.
Таблица 1 – Меры безопасности и контроля качества при производстве ВВИГ [4]
|
Производственный этап |
Требования к этапу, критические для обеспечения качества/безопасности ВВИГ |
|
Учреждение по заготовке крови (лицензирование и проверяются национальным регулирующим органом; контроль оборудования, фракционирующего плазму) |
|
|
Скрининг донор крови и плазмы |
Эпидемиологический надзор за населением, идентификация доноров, конфиденциальное анкетирование кандидатов в доноры на наличие факторов риска, анализ их медицинских документов и анкеты |
|
Процедура сбора крови/плазмы |
Контроль длительности процедуры забора крови у доноров, смешивания с растворами, предотвращающими коагуляцию, температуры от момента забора крови до ее направления в блок переработки и др. параметров процесса, определенных нормативным документом |
|
Тестирование донора на вирусоносительство перед забором крови |
Выявление антител к ВИЧ 1 и 2, вирусам гепатитов А, В и С, HBsAg, парвовируса В19. Исследование должно проводится индивидуально или минипулами, использанные методы должны быть валидированы |
|
Другие тесты у доноров |
Тестирование на изоагглютинины к антигенам А, В, D, не использование крови доноров с высокими титрами антител к этим антигенам |
|
Переработка крови/плазмы |
Должна использоваться плазма, замороженная в течение 24–72 ч после забора |
|
Замораживание и хранение плазма |
Должен использовать быстрый способ замораживания плазмы, в процессе ее хранения температура не должна меняться |
|
Транспортировка плазмы |
Во время транспортировки должен вестись постоянный мониторинг температуры хранения плазмы с записью соответствующим оборудованием. Температура хранения при транспортировке должна быть минус 20 0С или менее |
|
Предприятия по фракционированию плазмы крови (лицензирование и инспекция национальным регулирующим органом) |
|
|
Тестирование мини-пулов |
Используются технологии амплификации нуклеиновых кислот. Определяется нуклеиновая кислота ВИЧ 1 и 2, вирусов гепатитов А, В и С, парвовируса В19 |
|
Тестирование производственного пула |
Антитела на ВИЧ 1 и 2, вирус гепатита С, HBsAg (обязательно); РНК вируса гепатита С (обязательно в Европе). Исследование нуклеиновых кислот других вирусов – в соответствии с регулирующими документами |
|
Предприятие по фракционированию плазмы |
Должно быть разработано, построено и функционировать в соответствии с GMP |
|
Этапы очистки белков и инактивация вирусов |
Все процессы должны быть валидированы, все операции должны выполняться в соответствии с утвержденной СОП |
|
Стерилизующая фильтрация и асептическое заполнение упаковок |
То же |
|
Лиофильное высушивание (когда необходимо) |
То же |
|
Проверка конечного продукта |
Все операции должны выполняться в соответствии с утвержденной СОП |
Устранение недостатков, характерных для ВВИГ на основе интактного IgG, проводилось путем более тщательной очистки препарата от агрегатов иммуноглобулинов, протеаз, плазмина, плазминогена, активатора прекалликреина, примесей IgA и IgM. Для этого в технологический процесс их получения вводились дополнительные этапы фракционирования (спиртовое по Кону, использующие хроматографическое разделение компонентов крови, нанофильтрацию и др.)
Донюш [3] выделяет 4 поколения ВВИГ:
препараты первого поколения – начало 1970-х гг., это энзиматически и химически модифицированные IgG, не имевшие функционального Fc-фрагмента;
препараты второго поколения – конец 1970-х гг., включали полностью интактную молекулу IgG с активной Fc-функцией и могли применяться не только с целью заместительной, но и иммуномодулирующей терапии. Однако степень их очистки оставалась низкой, они содержали IgA в количествах, вызывающих анафилактические реакции при внутривенном введении, показатель Fc-функции не превышал 70–75%;
препараты третьего поколения создавались в середине-конце 1980-х гг., характеризовались высокой чистотой и полной активностью Fc-фрагмента, высокой степенью вирусной безопасности, достигаемой многоступенчатым процессом производства. Выпускались в жидком виде и могли храниться при температуре 2–8 0С;
препараты четвертого поколения – препараты, удовлетворяющие более жестким требованиям вирусной безопасности и физиологического распределения IgG по подклассам. Разработаны в 1990-х гг. и широко используются в настоящее время. Имеют высокую чистоту IgG с нормальным распределением по подклассам, содержание мономеров и димеров более 95%. Активность Fc-фрагмента молекулы IgG приближается к 100%.
Препараты четвертого поколения получают используя многоступенчатую схему инактивации вирусов, включающую не менее двух самостоятельных методов (сольвент-детергентная обработка + инкубация при низких значениях рН или пастеризация в сочетании с обработкой полиэтиленгликолем). Препараты выпускают в жидком виде, допускается хранение при комнатной температуре. В качестве стабилизаторов ВВИГ четвертого поколения используются вещества, безопасные для пациентов с нарушением углеводного обмена и дисфункцией почек, 10% растворы ВВИГ позволяют снизить объемную нагрузку на пациента [3].
Учитывая, что степень очистки IgG в препаратах четвертого поколения приближается к 100%, их можно считать пределом развития всего направления получения ВВИГ из плазмы крови доноров. Отдельные усовершенствования будут касаться:
повышения эффективности технологий получения и клинического применения ВВИГ (препарат для подкожного применения, комбинации различных иммуноглобулинов в препарате и др.);
эффективности выделения минорных белков плазмы;
вирусной безопасности препаратов крови;
способов очистки препаратов крови от примесей компонентов, которые раньше не считали способными влиять на результат клинического применения (растворимые молекулы CD4, CD8, HLA, следовые количества факторов свертывания крови VIII, IX, X, XI, XII и др.).
В таблице 2 обобщены данные по связи осложнений после введения пациенту ВВИГ с наличием отдельных контаминантов в препарате.
Таблица 2 – Связь осложнений, вызванных введением пациенту ВВИГ, с наличием в препарате контаминантов*
|
Осложнение |
Контаминант |
|
Озноб, тахикардия, гипотензия, кожная сыпь, лихорадка, шок, тромбогеморрагические осложнения |
Агрегаты иммуноглобулинов |
|
Лихорадка, тошнота, гипотензия |
Димеры IgG, образовавшиеся при хранении препарата |
|
Анафилактическая реакция у пациентов с врожденными нарушениями антителообразования |
IgA и IgM |
|
Тромбогеморрагические осложнения |
Факторы свертываемости крови VIII, IX, X, XI и XII |
|
Иммуносупрессия |
Антитела к интерлейкинам и их рецепторам |
|
То же |
Растворимые антигены CD4, CD8 и HLA II класса |
|
Гемолитическая анемия |
АВ-антитела |
|
Тромбогеморрагические осложнения |
Активаторы прекалликреина |
|
Крапивница |
Антитела к белкам плазмы крови |
|
Снижение терапевтической активности ВВИГ |
Фрагментация IgG сериновыми протеазами плазмы крови (калликреин и плазмин) |
|
То же |
Ненормальное распределение IgG по подклассам** |
|
То же |
IgG с изменённой структурой и конформацией, вызванных воздействием физических и химических факторов, использованных для инактивации вирусов |
|
Нарушения зрения (размытость зрительного поля, потеря резкости и др.), чувство покалывания кожи, гиперкалиемия |
Глицин (включается в препарат как стабилизатор) |
|
Аллергические реакции |
Метиленовая синь (используется при очистке плазмы крови от вирусов) |
*По работам [3, 4, 6, 8].
**Если это не препарат определенного терапевтического назначения. Например, ВВИГ, содержащие повышенное содержание IgG3-антител, имеют выраженную вируснейтрализующую активность [3].
Технологии получения ВВИГ и других компонентов крови, основанные на использовании криообедненной плазмы крови и осаждения белков крови этанолом при низких значения рН, показаны на рисунке 3.
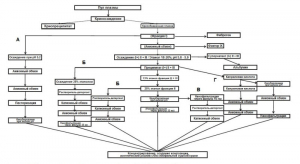 А, Б, В и Г – технологии, описанные в работах Bertolini J. [10], Teschner W. et al. [11], Terpstra FG. et al. [12] и Stucki M. еt al. [13] соответственно. Рисунок 3. Основные технологии, используемые для получения коммерческого ВВИГ и других компонентов крови из плазмы человека [4]
А, Б, В и Г – технологии, описанные в работах Bertolini J. [10], Teschner W. et al. [11], Terpstra FG. et al. [12] и Stucki M. еt al. [13] соответственно. Рисунок 3. Основные технологии, используемые для получения коммерческого ВВИГ и других компонентов крови из плазмы человека [4]

Получение коммерческого ВВИГ и других компонентов крови из плазмы человека по данной технологии начинается с отделения от нее так называемого «криопреципитата» (см. выше). Для этого замороженную плазму оттаивают в контролируемых условиях, затем центрифугируют с помощью центрифуг непрерывного действия (при температуре 2–3 0С). Полученный криосупернатант («cryosupernatant» или «cryo-poor plasma») используют для получения различных препаратов крови. Для выделения белков протромбинового комплекса, антитромбина или C1-ингибитора системы комплемента крови используют хроматографические методы. Для осаждения фибриногена супернатант может подвергаться первой этанольной преципитации (8% этанола при нейтральном значении pH). Для выделения ВВИГ используют три или четыре этапа осаждения этанолом при низких значения рН (осаждение по Кону) и обработку пепсином в низких концентрациях – при этом получают так называемую фракцию II (fraction II). Для уменьшения антикомплементарной активности ВВИГ и более глубокой очистки от IgА, осаждение этанолом может быть дополнено комбинацией этапов хроматографической очистки на катионных и анионных обменниках (с заменой третьего и четвертого осаждения по Кону). Выход фракции II при таком способе очистки плазмы составляет 3–6 г/л, в зависимости от исходного количества IgG в плазме и использованных методов его преципитации и последующей очистки [4, 7].
Стандартная технология производства позволяет из 1 литра плазмы получить до 2,5 упаковок альбумина 10%; до 3,5 упаковок иммуноглобулина для внутривенного введения 5%; и около 200–250 МЕ фактора VIII [9].
В тоже время данная технология слишком «груба» для выделения минорных белков плазмы крови. Поэтому к настоящему времени разработаны и используются в промышленном масштабе более эффективные технологии, заменяющие осаждение по Кону многоэтапным разделением белковых фракций крови аффинной хроматографией, например, «Plasma Protein Purification System (PPPSTM)». Суть таких технологий заключается в последовательном пропускании раствора, содержащего белки плазмы крови, через колонки с адсорбентом, включающим лиганд, специфически связывающий целевой белок, в то время как другие белки, липиды, липопротеины, углеводы, ДНК, пигменты, проходят через колонку не задерживаясь [14].
Основной тенденцией в стабилизации препаратов ВВИГ в настоящее время считается использование высокой концентрации IgG в препарате (100 мг/ мл по белку); слабокислой среды (рН 4,5–5,5); включение в лекарственную форму стабилизаторов, таких как полиолы (сорбит), сахара (мальтоза, глюкоза), или аминокислоты (глицин, пролин, изолейцин); отсутствие в препаратах хлорида натрия и сахарозы; осмолярность, близкая к физиологической; отсутствие консервантов и антибиотиков [6, 15].
Требования к свойствам ВВИГ, следующие [3]:
- они должны иметь оптимальный спектр антител в соответствии с инфицированностью населения (более 1000 доноров);
- обладать доказанной эффективностью (с помощью контролируемых клинических исследований);
- распределения IgG по подклассам должно соответствовать их содержанию в плазме крови;
- для каждой партии препарата должен быть задекларирован титр антител;
- макроагрегаты должны составлять менее 1% общего содержания IgG;
- антикомплементарная активность не должна превышать 1,0 СН50/1 мг белка протеина;
- гемолизины не должны содержаться в препарате, титр АВ-антител должен быть менее 1:8;
- активаторы прекалликреина, консерванты, активированные ферменты, токсические вещества не должны присутствовать в препарате;
- если предусмотрено применение у пациентов с врожденным дефицитом IgA; содержание IgA должно быть минимальным;
- высокая противовирусная очистка.
Иммуноглобулины для подкожного введения (subcutaneous immunoglobulin, SCIG). Возможность получения IgG с высокой степенью очистки позволила в последнее десятилетие вернуться к практике их подкожного введения, оказавшейся неудачной в 1940–1950-е гг. из-за большого количества реакций на балластные компоненты иммуноглобулинов, получаемых по технологиям того времени. Такие иммуноглобулины в основном используются для лечения пациентов с врожденными нарушениями антителообразования (низкий уровень IgA), с повышенным содержанием в сыворотке крови воспалительных маркеров, флебитами, заболеваниями почек и другой патологией, создающей условия для осложнения при введении ВВИГ [16]. В настоящее время за рубежом в клинической практике используется не менее 6 SCIG. Сравнение свойств SCIG с аналогичными свойствами ВВИГ приведено в таблице 3.
Таблица 3 – Сравнений свойств SCIG и ВВИГ [17]
|
Свойство |
ВВИГ |
SCIG |
|
Стабильность поддержания уровня IgG в сыворотке крови |
Невозможно |
Возможно |
|
Возможность пиков концентраций IgG в сыворотке крови |
Возможно |
Невозможно |
|
Защита от возбудителей инфекционных болезней |
Возможна |
Возможна |
|
Доступ к венам |
Нужен |
Не нужен |
|
Продолжительность инфузии |
Несколько часов |
Один час или меньше |
|
Частота инфузий |
Каждые 2–4 недели |
Обычно одно введение в неделю |
|
Возможность домашнего введения |
Возможно, но мало кто из пациентов пользуется такой возможностью |
Возможно, большинство пациентов предпочитает именно такое введение препарата |
Улучшение проницаемости внеклеточного матрикса для IgG в SCIG добиваются добавлением рекомбинантной гиалуронидазы человека (recombinant human hyaluronidase, rHuPH20), что позволяет сократить расход препарата на курс лечения пациента и добиться более высокого уровня антител в плазме крови [18]. По совокупности свойств и благодаря более простому применению в клинике, SCIG, особенно препараты с rHuPH20, способны потеснить ВВИГ на рынке фармацевтических препаратов.
Вирусная безопасность препаратов крови. Существует противоречие между требованиями к качеству и эффективности ВВИГ, и его безопасностью. Чтобы ВВИГ был эффективным и соответствовал критериям качества, плазма должна быть получена от как можно большего количества доноров, более 1000. Но чем больше донаций использовано для получения плазмы, тем больше риск того, что она будет инфицирована опасными для человека вирусами. Когда ВВИГ начали применять в клинической практике, считалось, что если IgG фракционируют холодным этанолом, то это обеспечивает вирусную безопасность полученных препаратов. Но эти надежды не оправдались, вирусы, особенно возбудитель гепатита С, продолжали находить в крупных партиях ВВИГ [19].
Почти все мировые производители препаратов крови сталкивались с проблемой вирусной контаминации своих продуктов [20]. В настоящее время решение этой проблемы осуществляется в двух направлениях: адекватная проверка исходного сырья и применение все более совершенных технологий инактивации вирусов. Тем не менее, проблема инфицирования больших пулов плазмы и как следствие, получаемых из них препаратов, осталась. В таблице 4 приведены сведения об основных способах, используемых для инактивации вирусов в препаратах крови.
Таблица 4 – Способы, используемые для инактивации вирусов в препаратах крови [4, 9, 20–22]
|
Способ |
Суть способа |
Ограничения способа |
|
Пастеризация жидкого продукта |
Нагревание продукта при температуре 60 0С в жидком состоянии 10–12 ч |
Риск заражения вирусами гепатитов В и С существует при использовании пастеризованных концентратов; необходимость использования больших концентраций протекторов, в основном углеводов с различными добавками, для защиты лабильных белков плазмы от денатурации. Одновременно они стабилизируют и вирусы |
|
Химическая инактивация вируса в жидком продукте |
Основан на способности химических веществ разрушать липидную оболочку вирусов. Использование растворителей (solvent-detergent Method; SD-метод) для удаления липидной оболочки |
Применение ограничено из-за лабильности белков плазмы крови. Способ применим для инактивации вирусов, имеющих липидную оболочку. Малоэффективен для инактивиции вирусов, вызывающих гепатит А или парвовирусную инфекцию. SD-метод запрещен в США из-за ассоциации с развитием у отдельных пациентов тромбозов. Может приводить к снижению биологической активности препарата и к образованию аутоантител. Например, метод, использующий метиленовую синь, снижал активность фибриногена на 65%; фактора крови VIII на 67% |
|
Химическая обработка + ультрафильтрация продукта |
Присоединение к химической обработке метода ультрафильтрации |
Необходимость удаления продуктов распада вирусов требует введение дополнительных этапов очистки препарата крови |
|
Ультрафиолет + химические вещества |
Плазму и ее препараты подвергают облучению светом в ультрафиолетовом диапазаоне в присутствии малых концентраций химических веществ – красителей (метиленовый синий и др.) |
Способ применим для инактивации вирусов, имеющих липидную оболочку. Малоэффективен для инактивиции вирусов, вызывающих гепатит А или парвовирусную инфекцию. Удаление продуктов распада вирусов требует дополнительных этапов. Возможна частичная денатурация терапевтических белков и образование к ним аутоантител |
|
Обработка лиофилизата паром |
Лиофилизат подвергают обработке горячим паром в закрытой системе, заполненной инертным газом под давлением в течение 1–10 ч |
В некоторых препаратах обнаруживали вирус гепатита В |
|
Сухой прогрев лиофилизата |
Инактивация вирусов происходит при нагреве лиофилизата при температуре 68 0С в течение 32–60 ч |
В концентрированных негомогенных препаратах сохранялись вирусы гепатитов В, С и ВИЧ |
|
Жесткая термообработка лиофилизата |
Прогрев до температуры 80 0С в течение 72 ч |
Требуются протекторы, возможна частичная денатурация белков. Возможно сохранение в препарате вирусов, вызывающих гепатит А или парвовирусную инфекцию |
|
Хроматография (гельфильтрация, афинная и ионообменная хроматография) |
Разделение происходит в результате межмолекулярного взаимодействия белков оболочки вируса с сорбентом |
Хроматографические способы малоэффективны при удалении безоболочечных вирусов. Обычно их применяют для получения из плазмы крови минорных белков |
|
Ультрафильтрация |
Механическое отделение вирусов за счет их больших размеров, чем у белков плазмы. Способ эффективен для удаления оболочечных и безоболочечных вирусов. Фильтры с размером пор 15–20 нм, позволяют отделить вирус гепатита A и вирус B19 от фактора IX |
Используется как этап многостадийных методов очистки крови от вирусов. Размеры частиц вируса, вызывающего гепатит A, находятся в пределах 25–30 нм; вируса B19 – в пределах 18-26 нм; вируса, вызывающего гепатит С, 28-30 нм; размеры вирусов герпетического семейства в пределах 120-300 нм; капсид ВИЧ – 100–120 нм |
|
Ультракороткие импульсы лазерного излучения (длина волны 425 нм) |
Разрушение вирусов в жидкой среде. Способ эффективен для удаления оболочечных и безоболочечных вирусов |
Нет данных |
По мнению Панова [20], вирусная безопасность продуктов крови обеспечивается тремя непременными факторами: отбором доноров, тестированием стадии донации и пулов плазмы, инактивацией и удалением вирусов в процессе производства препаратов крови. Процедуры, обеспечивающие выполнение этих факторов, должны соответствовать правилам GMP. Максимальная степень вирусной безопасности может быть достигнута путем комбинации всех трех факторов. Обязательной является процедура валидации способов инактивации и удаления вирусов.
Методы удаления вирусов из препаратов крови постоянно совершенствуются. Сравнительно недавно разработан способ, использующий ультракороткие импульсы лазерного излучения, по утверждению разработчиков, способный инактивировать вирусы обоих типов [21]. Однако современные технологические процессы производства препаратов из плазмы крови доноров должны включать, как минимум, две эффективных стадии вирусной редукции, сочетающиеся со стадиями очистки самого препарата от других компонентов плазмы крови. Причем одна из таких стадий должна эффективно удалять из препарата безоболочечные вирусы [9]. На рисунке 4 показана типовая многостадийная схема производственного процесса получения фактора крови IX. Этапы сольвент-детергентной обработки и ультрафильтрации используются для исключения вирусной контаминации препарата. Этап ультрафильтрации предназначен для удаления безоболочечных вирусов.
Плазма крови человека
↓
Криопреципитация белков
↓
DEAE-Sephadex A-50-анион-обменная хроматография
↓
Сольвент-детергентная обработка
↓
DEAE-Toyoperl 650M-анион-обменная хроматография
↓
Гепарин-Сефароза 6FF-афинная хроматография
↓
Ультрафильтрация (используются фильтры Viresolve NFP)
↓
Асептическое заполнение флаконов
↓
Лиофилизация препарата
Рисунок 4. Схема получения высокоочищенного и свободного от вирусной контаминации препарата фактора крови IX [22]
В целом же данные, приведенные в таблице 4, показывают, что до настоящего времени не разработаны единые для всех производителей препаратов крови технологические приемы, позволяющие гарантировать их вирусную безопасность на 100 %. Любым их сочетанием достигается только «максимальная степень вирусной безопасности» (см. [4, 20]). Риск контаминирования вирусами препаратов крови возрастает при масштабировании производства, использовании концентрированных препаратов и расширении круга производителей.
Прионовая безопасность препаратов крови. Прионный белок (PrPC) – относительно простой мономерный белок с ММ 27–30 кДа, обладающий активностью супероксиддисмутазы. Синтезируется в нейронах в виде предшественника и подвергается в последующем протеолитическому созреванию и пострансляционной модификации. Участвует в процессе клеточного распознавания или передачи нервного импульса через рецепторы. Прионы (англ. prion от protein — «белок» и infection — «инфекция», термин предложен в 1982 г. Stanley B. Prusiner) — белковые инфекционные частицы, изоформы прионных белков (PrPSc), имеющие аномальную структуру, способную катализировать конформационное превращение гомологичного ему нормального клеточного прионного белка в себе подобный (прион) (рисунок 5).
PrPC включает три альфа-спирали (F, B и C; обозначены зеленым цветом) и две короткие бета-складки (обозначены красным цветом). PrPSc состоит в основном из бета-складчатых структур, оставшиеся B и С альфа-спирали имеют измененную конформацию. Аминокислотные последовательности PrPC и PrPSc идентичны.
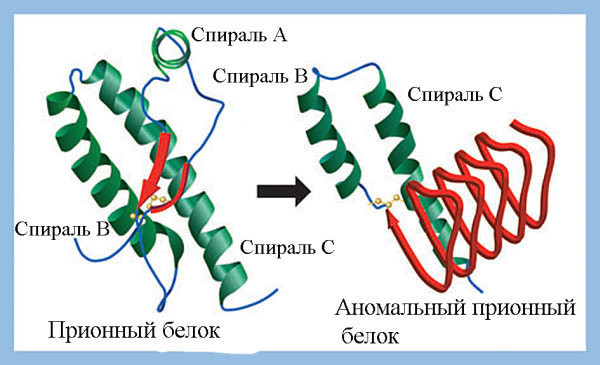
Рисунок 5. Третичная структура «нормального» (PrPC) и «аномального» (PrPSc) прионного белка [23].
Каждая молекула PrPSc, инфицировавшего клетку, конвертирует одну молекулу PrPС в себе подобную. Две PrPSc молекулы формируют из двух молекул PrPС две PrPSc. Запускается цепная реакция, в ходе которой в ЦНС образуется огромное количество утративших растворимость и неправильно свёрнутых молекул полимеризовавшихся прионных белков, формируются амилоидные бляшки, разрушающие нормальную структуру ткани, что приводит к развитию у инфицированного человека или животного нейродегенеративного заболевания [24, 23].
Прионовые болезни – это принципиальная иная эпидемическая проблема, чем те, с которыми сталкивались эпидемиологи при противодействии вспышкам чумы, дифтерии, гриппа, или, даже, пандемиям ВИЧ-инфекции и сывороточных гепатитов. Длительный период времени между эпизоотиями прионовых болезней среди животных и эпидемиями среди людей, вызванными мясом, инфицированным прионами, затрудняет выявление конкретного источника заражения. Например, ретроспективно было установлено, что временной интервал между пиком вспышки варианта болезни Крейтцфельдта–Якоба (variant Creutzfeldt–Jakob disease, vCJD) среди людей, последовавшей за эпизоотией бешенства коров в Великобритании, составил 9 лет [23]. Количество людей, инфицированных прионами, но находящихся в инкубационном периоде болезни, неизвестно. В Соединенном Королевстве один такой больной может приходиться на 2500–4000 жителей [26, 27]. Способы, используемые для инактивации вирусов в препаратах крови, сложно адаптировать для инактивации прионов. Прионы проходят через фильтры с порами 25–50 нм в диаметре; сохраняют стабильность своей структуры даже при температуре 90 0С. Благодаря гидрофобности они имеют выраженную тенденцию к агрегации инфекционных единиц между собой и с клеточными белками и структурами. Их инактивация возможна при автоклавировании при температуре 135 0С в течение 30 мин [24], что неприменимо для обеспечения прионовой безопасности препаратов крови. Недостаточно оцененную опасность для здоровья людей представляет накопление знаний, позволяющих синтезировать прионы de novo и распространять их путем искусственного заражения сырья, используемого для получения препаратов крови [28].
Проблема прионовой безопасности уже дала о себе знать производителям препаратов крови. Известно о пяти случаях заражения людей прионами, вызывающими vCJD. В четырех случаях заражение вызвала эритроцитарная масса, в одном – препарат фактора свертываемости крови VIII. У доноров симптомы vCJD обнаружены в период от 1 года и 4 мес до 3 лет и 4 мес после забора у них крови [23, 29–31] (рисунок 6).
Прионовая безопасность препаратов крови может быть обеспечена тщательным анализом анамнеза донора, обнаружением PrPSc в крови доноров и пулах плазмы, использованием технологий очистки плазмы крови, исключающих наличие прионов в конечном продукте.
Исследование анамнеза донора должно включать определение географии его проживания за 10-летний период и наличие за этот период времени на этой территории вспышек прионовых болезней среди животных.
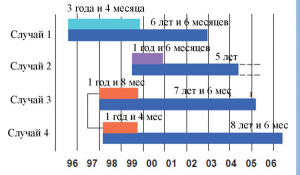 Верхняя линия – временной интервал, в течение которого появились симптомы vCJD у доноров плазмы, нижняя – у ее реципиентов. Случаи 3 и 4 вызваны заражением vCJD от одного донора. Рисунок 6. Временные интервалы развития симптомов vCJD у доноров плазмы и реципиентов препаратов крови, полученных из плазмы крови инфицированного прионами человека [23].
Верхняя линия – временной интервал, в течение которого появились симптомы vCJD у доноров плазмы, нижняя – у ее реципиентов. Случаи 3 и 4 вызваны заражением vCJD от одного донора. Рисунок 6. Временные интервалы развития симптомов vCJD у доноров плазмы и реципиентов препаратов крови, полученных из плазмы крови инфицированного прионами человека [23].
Для обнаружения прионов в образцах жидкостей и тканей человека и в препаратах крови используют систему циклической амплификации неправильно скрученного белка (Protein Misfolding Cyclic Amplification, PMCA). Система основана на том же принципе формирования аномальной структуры прионового белка, который используют прионы для своего размножения в условиях in vivo [32]. Начальной матрицей для амплификации служит PrPSc, содержащийся в пробе. Ее разводят избытком PrPC, используемого в качестве субстрата, и инкубируют в условиях, наиболее оптимальных для образования агрегатов прионового белка, изменившего свою структуру на аномальную, т.е. способную катализировать конформационное превращение гомологичного ему нормального клеточного прионного белка. Такие формы белка, образовавшиеся в условиях in vitro, называют PrPres. Агрегаты разрушают ультразвуком, цикл многократно повторяют [25]. Для повышения чувствительности и воспроизводимости PMCA при обнаружении PrPSc в крови пациента исследователи идут по двум направлениям. Подбирают наиболее специфический субстрат для амплификации PrPSc и повышают чувствительность способов обнаружения амплификатов, т.е. PrPres.
По первому направлению в 2014 г. было показано, что для обнаружения PrPSc в крови человека наилучшие результаты дает использование овечьего нормального прионного белка Q171 PrP (замена Q171R в белке PrP овец препятствует образованию PrPSc). Следовательно, при выборе субстрата гомология между ним и матрицей является не критичным условием эффективности амплификации PrPres [33].
Работы по второму направлению вывели к использованию в 2011 г. для обнаружения прионов технологи Surround Optical Fiber Immunoassay (SOFIA) («оптический иммунологический анализ прилежащих волокон»). После амплификации с концентрированием всех PrPres, содержащихся в образце, их помечают флуоресцентным красителем со специфическими антителами и загружают в микрокапиллярную трубку. Трубку помещают в специальный аппарат, где она оказывается полностью окружённой оптическими волокнами и весь свет, испускаемый волокнами на трубку, поглощается красителем, предварительно возбуждённым лазером. Эта технология позволяет обнаружить PrPSc даже после небольшого количества циклов перехода прионного белка в прионную форму, что, во-первых, снижает возможность искажения результата артефактами эксперимента, и, во-вторых, сокращает продолжительность исследования [34, 36].
Для повышения точности и специфичности исследования разработаны стандартные образцы прионов разных видов животных [36].
Технологии очистки плазмы крови, исключающие наличие прионов в конечном продукте, несовершенны. На рынок выпущен только один препарат ВВИГ, имеющий маркировку Управления по санитарному надзору за качеством пищевых продуктов и медикаментов Министерства здравоохранения и социальных служб США (Food and Drug Administration, FDA) об удалении прионовых белков [3].
Критерии качества препаратов крови. Их определяет национальная фармакопея. Методы, используемые при оценке качества препаратов крови должны быть валидированы. Показатели качества ВВИГ приведены в таблице 5.
Таблица 5 – Показатели качества ВВИГ [4]
|
Показатель |
Норма |
|
Растворимость лиофильно-высушенного продукта |
Менее 10 мин при температуре 20 0С |
|
Прозрачность |
Полная, не должно быть взвешенных частиц и невидимых частиц установленного размера |
|
рН |
4,0–4,5 или 6,8–7,4 |
|
Осмотическое давление, мОсм?моль/кг |
≥ 240а |
|
Общий белок, г/л |
Не менее 30а |
|
Содержание гамма-глобулина, % |
≥ 95а |
|
Димер + мономер, % |
≥ 90а |
|
Агрегаты, % |
≤ 3а |
|
Вспомогательные вещества (например, натрия хлорид, глицин, мальтоза, маннит, сахароза и т.п.) |
В соответствии с нормативным документом |
|
Вирус-инактивирующие агенты (сольвенты, детергенты, три(n-бутил)фосфат, мкг/мл |
≤ 10 или в соответствии с нормативным документом |
|
Полисорбат 80, мкг/мл |
≤ 100 или в соответствии с нормативным документом |
|
Титр гемагглютинина Анти А |
Негативный при разведении 1/64 (при концентрации белка 3%)а |
|
Титр гемагглютинина Анти В |
Негативный при разведении 1/64 (при концентрации белка 3%)а |
|
Титр гемагглютинина Анти D |
≤ 1а |
|
IgА |
В соответствии с нормативным документом. Максимальное количество должно быть указано на упаковке препаратаа |
|
Активатор прекалликреина (prekallikrein activator, PKA), МЕ/мл |
Менее 35 (при концентрации белка 3%)а |
|
Тест на идентичность человеческому происхождению |
Положительныйа |
|
Антикомплементарная активность, CH50⁄мг IgG |
≤ 1а |
|
Целостность Fc-фрагмента |
Рутинного способа анализа не существует, используют применяемый при производстве иммуноглобулина |
|
Титры специфических антитела |
В соответствии с регистрационным удостоверением лекарственного средстваб |
|
HBsAg |
Отсутствует а |
|
Антитела к ВИЧ 1 и 2 |
Отсутствуют |
|
Антитела к HCV |
Отсутствуют |
|
Бактериальная стерильность |
Стерилен а |
|
Тест на эндотоксин, МЕ/мл |
Менее 0,5 (при содержании белка 5%), менее 1,0 (содержание белка 5–10%) а |
|
Anti-HBs, МЕ/мл |
≥ 0,5 а |
|
Тест на пирогенность (LAL-тест) |
Апирогенен |
|
а – European Pharmacopoeia specification; б – при концентрации белка в препарате 50 г/л он должен содержать как минимум два специфических антитела (одно к возбудителю вирусной инфекции, другое – к бактериальной) в концентрации, по крайней мере, в три раза более высокой, чем в стандартных препаратах ВВИГ |
|
Стандарты ВОЗ и референс-препараты ВВИГ и других препаратов крови, европейские производители получают из Национального института биологических стандартов и контроля (National Institute for Biological Standards and Control, NIBSC; Potters Bar, UK), Центра оценки и исследований биологических препаратов Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (Center for Biologics Evaluation and Research, Food and Drug Administration; CBER⁄FDA, Bethesda, MD) или из Европейского директората по качеству лекарственных средств и здравоохранения (European Directorate for the Quality of Medicines and HealthCare; EDQM, Strasbourg, France). Каталоги международных стандартов доступны на сайтах NIBSC и ВОЗ.
В то же время, современная концепция получения ВВИГ как пула IgG с высокой степенью очистки, выделенного из плазмы тысяч доноров (ВВИГ четвертого поколения), содержит ряд ограничений, неразрешимых в ее собственных рамках.
Сходство различных партий ВВИГ по физико-химическим свойствам, не означает, что сходным будет терапевтический эффект после их введения пациенту. Даже по приблизительным оценкам в стандартных поливалентных ВВИГ могут определяться антитела к 17 бактериальным антигенам, 21 антигену вирусов, 6 антигенам грибов и простейших [8, 37]. Невозможно добиться воспроизводимых результатов, экспериментируя системой, в которой действуют по неизвестным закономерностям не менее 54 неизвестных факторов.
Открыта и проблема вирусной безопасности препаратов крови. В соответствии с рекомендациями ВОЗ и Европейского агентства по оценке медицинских продуктов, стадия очистки от вирусов считается эффективной, если она обеспечивает редукцию патогенов в 104 раз и более [9]. Однако снижение количества вирусных частиц в конечном продукте не означает их отсутствие. Увеличение количества стадий очистки одновременно снижает производительность технологического процесса и удорожает конечный продукт. Существует проблема качества самих тест-систем.
Иммунный ответ на введение ВВИГ трудно прогнозировать. Высокие дозы ВВИГ способны угнетать продукцию интерлейкинов (ИЛ) и снижать уровень экспрессии рецепторов к ИЛ2 вследствие наличия в препарате соответствующих антител, а также блокировать Fc-рецепторы при активных Fc-фрагментах IgG. Содержание в препаратах ВВИГ ряда биологически активных протеинов, например, растворимых антигенов CD4, CD8 и антигена главного комплекса гистосовместимости (HLA) II класса, также оказывает влияние на реализацию физиологического иммунного ответа на антигенный раздражитель (см. табл. 2) [4].
С позиций доказательной медицины в настоящее время подвергается сомнению терапевтическая эффективность ВВИГ при терапии сепсиса и септического шока. Многолетняя клиническая практика и экспериментальные работы показали, что только введение ВВИГ, обогащенных IgM и IgA, повышает выживаемость пациентов. Однако IgA при случайном введении пациенту с врожденным дефицитом IgA, вызывает анафилактические реакции. В многоцентровом исследовании не обнаружено снижения летальности при использовании стандартных ВВИГ у кардиохирургических больных с тяжелой послеоперационной системной воспалительной реакцией, считавшейся показанием к их применению [3].
Не устранена проблема наличия в препаратах растворимых молекул CD4, CD8 и HLA II и I и антител к ним, способных влиять на распознавание антигена лимфоцитами; а также тромбина, факторов крови VIII, IX, X, XI и XII в количествах, активирующих свертывание крови и приводящих к образованию множественных тромбов у пациента (см. табл. 2).
Нерешенность этих проблем приводит к неопределенности результатов клинического применения ВВИГ четвертого поколения и требует их дальнейшего совершенствования, в том числе за счет расширения оцениваемых показателей качества. Одновременно можно предположить появление на рынке препаратов иммуноглобулинов иного типа: полученных генно-инженерным путем (без использования плазмы крови человека), поэтому хорошо охарактеризованных по молекулярному составу и обладающих высокой избирательностью в отношении мишеней воздействия.
2. Гетерологичные иммуноглобулины
Гетерологичные иммуноглобулины представляют собой лекарственные средства, содержащие очищенные иммуноглобулины или их фрагменты, полученные из сыворотки или плазмы животных различных видов (чаще лошадей), иммунизированных соответствующими антигенами. Препараты содержат в высокой концентрации специфические антитела, нейтрализующие или связывающие антигены, использованные для иммунизации животных [6].
Технологии получения иммуноглобулина из гетерологичных сывороток, по основным этапам фракционирования практически ничем не отличаются от получения иммуноглобулинов из сыворотки человека. Их совершенствование идет в направлении повышения специфичности иммуноглобулинов, степени очистки от балластных примесей и вирусной безопасности. Критерии качества гетерологичных иммуноглобулинов определяют национальные фармакопеи [38, 39]. В тоже время возможности технологий их получения имеют предел в развитии – гетерологичные иммуноглобулины всегда будут поликлональными, и их свойства будут различаться в зависимости от источника плазмы крови [40].
Тенденции в развитии способов получения таких препаратов, и, соответственно, методов контроля их качества, прослеживаются на примере получения гетерологичных иммуноглобулинов, предназначенных для лечения отравлений, вызванных зоотоксинами (укусы ядовитых змей и членистоногих; ранения, нанесенные ядовитыми морскими животными). Впервые антитоксическая сыворотка против яда змеи Vipera aspis (асписовая гадюка) получена из крови морских свинок в 1894 г. сотрудниками филиала Института Пастера (Pasteur Institute) в Сайгоне [41].
Яды животных представляют собой сложный объект для разработчиков специфических иммуноглобулинов из-за многокомпонентности их состава. Они состоят из смеси синергидно действующих биологически-активных веществ, среди которых токсин, определяющий клинику и тяжесть поражения, в процентном отношении занимает незначительную часть по отношению к другим компонентам [42]. Наработать антитоксические иммуноглобулины против зоотоксинов, используя человека в качестве донора плазмы, невозможно. Поэтому с 1894 г. это направление специфической профилактики и лечения поражений, вызванных укусами ядовитых животных, основывается на использовании иммуноглобулинов, полученных из плазмы животных различных видов.
Не более 20 % антител, вырабатывающихся иммунной системой животного в ответ на гипериммунизацию инактивированным змеиным ядом, специфичны к его компонентам. Из них менее чем 5% представлены нейтрализующими антителами, способными с различной афинностью (!) взаимодействовать с эпитопами токсина и блокировать его взаимодействие с рецепторами на поверхности клетки-мишени [40] (рисунок 7).
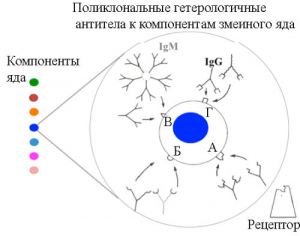 А, Б, В и Г – эпитопы молекулы токсина, «узнаваемые» поликлональными антителами. А – эпитоп в активном сайте токсина. Антитела, взаимодействуя с этим эпитопом, изменяют конформацию молекулы токсина и предотвращают его связывание с рецептором на поверхности клетки-мишени. Синим цветом обозначен наиболее токсичный компонент змеиного яда. Рисунок 7. Взаимодействие в условиях in vivo поликлональных гетерологичных антител с компонентами змеиного яда [40].
А, Б, В и Г – эпитопы молекулы токсина, «узнаваемые» поликлональными антителами. А – эпитоп в активном сайте токсина. Антитела, взаимодействуя с этим эпитопом, изменяют конформацию молекулы токсина и предотвращают его связывание с рецептором на поверхности клетки-мишени. Синим цветом обозначен наиболее токсичный компонент змеиного яда. Рисунок 7. Взаимодействие в условиях in vivo поликлональных гетерологичных антител с компонентами змеиного яда [40].
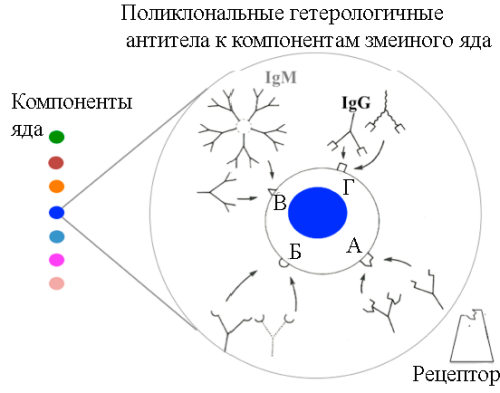
Многокомпонентность таких ядов и сложная конформационная структура молекул токсинов, ограничивают использование мышиных моноклональных антител (мАТ), специфичных к отдельным эпитопам токсина и являющихся для иммунной системы человека антигенами. Эти два обстоятельства вынуждают исследователей для повышения эффективности и безопасности иммуноглобулиновых препаратов разрабатывать коктейли из рекомбинантных антител, высокоафинных к эпитопам токсина [40].
При изучении зарубежной научной литературы по другому «опережающему объекту» – способам получения ботулинических антитоксинов, установлено, что в клинической практике США и Европы используются получаемые из лошадиной сыворотки три-, би- и моновалентные антитоксины, произведенные Sanofi Pasteur Limited, California Department of Public Health и CDC/USA по технология, разработанным еще в 1950–1960-х гг. Более новые работы описывают лишь частные усовершенствования известных способов получения иммуноглобулинов. Li et al. [43] описали способ гипериммунизации лошадей, позволяющий получать моноспецифические гипериммунные сыворотки к ботулиническим токсинам типов А и В. Усовершенствование способа получения специфических сывороток заключалось лишь в щадящей обработке ботулинического токсина формалином, что не выходит за рамки представлений о получении анатоксинов, сформировавшихся в начале XX в. В тоже время анализ научной литературы позволил обнаружить два направления, конкурирующих с технологией получения ботулинических антитоксинов из сыворотки животных: получение препаратов на основе моноклональных антитела различного типа и высокоаффинных к ботулиническим токсинам Fab-фрагментов IgG1 человека, полученных в дрожжах генно-инженерным путем [44, 45], т.е. тенденция их дальнейшего развития та же, что и у препаратов иммуноглобулинов, предназначенных для лечения отравлений, вызванных зоотоксинами.
Публикации, раскрывающие принципиально новые технологии получения препаратов антидифтерийных и антистолбнячных иммуноглобулинов из гетерологичных сывороток, не были найдены, это показывает исчерпание возможностей развития технологий получения гетерологичных иммуноглобулинов, и отражает общую тенденцию на рынке препаратов крови – замена ксеногенных компонентов на компоненты с установленными составом и структурой, полученных генно-инженерным путем.
3. Рекомбинантные препараты крови
Объективными причинами, способствующими развитию технологий получения рекомбинантных препаратов крови, являются недостаток донорской крови, риск передачи вирусных и прионных инфекций, высокая стоимость терапии, для которой используются такие препараты [3, 4, 23, 29, 46]. Перспективы промышленного производства препаратов крови с помощью технологий генной инженерии появились в 1980-е гг., когда были клонированы факторы крови IX и VIII и созданы системы их экспрессии в клеточных культурах млекопитающих [47, 48]. В настоящее время по технологиям генной инженерии производятся не менее 20 препаратов крови [46]. Их качество, эффективность и безопасность, прежде всего, зависят от использованных производителем систем экспрессии клонированных генов факторов крови. Крупные белки обычно синтезируют в клетках млекопитающих, способных выполнить гликозилирование молекулы белка[1] таким же образом, как это происходит в клетках человека. Возможности современных систем экспрессии генов белков крови, приведены в таблице 6.
Таблица 6 – Возможности современных систем экспрессии генов белков крови [46]
|
Система экспрессии |
Может быть использована |
Не может быть использована |
Особенности экспрессионной системы |
|
Esche-richia coli |
Для экспрессии простых полипетидов (инсулин, гормон роста человека, интерферон-альфа, фактор роста человека) |
Для экспрессии сложных гликопротеинов плазмы крови, обладающих большой ММ |
В E. coli не может быть достигнута правильная пострансляционная модификация крупной гликопротеиновой молекулы, что приводит к ее не физиологическому фолдингу и нарушению формирования дисульфидных связей между цепями. Такие молекулы не способны длительное время циркулировать в сыворотке крови человека, быстро агрегируются при хранении и утрачивают биологическую активность. Например, синтезированный в E. coli альфа-1-антитрипсин в доклинических исследованиях показал низкую стабильность in vitro и измененные фармакоркинетические свойства |
|
Дрожжи (Saccha-romyces cerevi-siae, Pichia pastoris, Hanse-nula polymor-pha) |
Для экспрессии интерферона-альфа, фактора роста человека (гликопротеины с ММ менее 60 кДа), альбумина и негликозилированных белков плазмы крови
|
То же |
Дрожжи могут выполнять ранние стадии N-гликозилирования в эндоплазматическом ретикулуме. Сложный олигосахаридный синтез и гликановое созревание (glycan maturation) им не доступны. Синтезированные белки гетерогенны и быстро выводятся из кровеносного русла |
|
Нит-чатые грибы |
Небольшой опыт использования для экспрессии генов белков крови |
Выполняет гликозилирование также как клетки млекопитающих, в отличие от клеток дрожжей не вызывают гиперманнозилирования белков (hypermannosylation) |
|
|
Клетки насекомых (баку-лови-русные экспресс-ионные систе-мы) |
Для экспрессии простых полипетидов |
Система не позволяет экспресс-сировать сложные гликопротеины плазмы крови |
Выполняют пострансляционные модификации молекул гликопротеина, такие как N- и O-гликозилирование или формирование дисульфидных связей и достигать правильного белкового фолдинга. Однако их механизм гликозилирования гетерологичного белка в основном не описан. Существует риск добавления к молекуле дополнительных сахаров, не характерных для человека |
|
Клетки млеко-пита-ющих CHO и BHK*
|
Для экспрессии большинства рекомбинантных белков плазмы крови
|
Нет данных |
Выполняет сложную пострансляционную модификацию и обеспечивают секрецию белков в среду в исходной форме. В настоящее время разработаны питательные среды, состоящие из компонентов, полученных генноинженерным путем и не содержащие ксеногенные компоненты |
|
Трансгенные животные |
Для получения антитромбина, альфа-1-антитрипсина, ингибитора С1-эстеразы, фибриногена, альбумина, мАТ, факторы крови FVIII и FIX |
То же |
Существуют риск прионной и вирусной контаминации препарата и возможность иммунных реакций на его введение, так как сложные посттрансляционные модификации (C-карбоксилирования, бета-гидроксилирования; N- и О-связанное гликозилирование; фосфорилирование ) экспрессируемого белка происходят видо- и тканеспецифическим образом, характерным для данного вида трансгенного животного, но не человека |
|
Транс-генные растения |
мАТ |
То же |
Трансгенные растения могут выполнять посттрансляционные модификации, в частности модификации N-гликанов, но их аппарат Гольджи не в состоянии синтезировать некоторые сахара и терминальные сиаловые кислоты, что может приводить к иммунным реакциям на введенный препарат |
* CHO (от сhinese hamster ovary – яичники китайского хомяка) и BHK (от baby hamster kidney – почки детенышей хомячка).
Лекарственные препараты на основе полноразмерных рекомбинантных факторов FVIII и FIX используются в клинической практике с начала 1990-х гг. Их получают на основе систем экспрессии генов FVIII и FIX в эукариотических клетках CHO и BHK, и с помощью трансгенных животных-продуцентов (молоко коз и свиней). Рекомбинантный FVIII, секретируемый клетками CHO в комплексе с рекомбинантным vWF (фактор фон Виллебрандта), выпущен под торговыми названиями Recombinate® и Bioclate®. Рекомбинантный FVIII, секретируемый клетками BHK в культуральную среду, содержащую природный vWF, получил торговые названия Kogenate® и Helixate® (см. табл. 6).
Различают три поколения лекарственных препаратов рекомбинантных факторов свертывания крови [49]:
первое поколение – лекарственная форма содержит человеческий сывороточный альбумин и контактируют с веществами животного происхождения в процессе производства;
второе поколение – лекарственная форма не содержит альбумина в качестве вспомогательного вещества;
третье поколение – контакт с веществами животного происхождения и компонентами донорской плазмы исключен во всем процессе производства.
К настоящему времени сформировалось четыре направления создания таких препаратов: полноразмерные рекомбинантные FVIII и FIXl; их делеционные варианты (для увеличения выхода продукта в экспрессионной системе убирается B-домен); пролонгированные производные (к молекуле фактора крови или его делеционного варианта ковалентно присоединяются группы полиэтиленгликоля); и гибридные факторы свертывания крови, ковалентно сшитые с доменом Fc иммуноглобулина человека G1 [48]. Присоединение к молекуле rFIX Fc-домена IgG1 увеличивает продолжительность циркулирования в кровеносном русле rFIX. Биоинженерные стратегии получения рекомбинантных факторов свертывания крови приведены в работе Peyvandi et al. [51].
Получение препаратов рекомбинантных факторов свертывания крови осуществляется в соответствии с правилами надлежащей лабораторной практики (Good Laboratory Practice) [50, 51]. По показателям активности синтезируемого продукта, физико-химических свойств продукта (обязательное требование – синтез рекомбинантного фактора крови в мономерной форме), производительности и стабильности, отбирается клеточная линия-продуцент. Клеточную линию с наиболее оптимальными показателями субклонируют и используют в качестве продуцента фактора свертывания крови.
Общей тенденцией в получении рекомбинантных белков крови является использование многоэтапного афинного и псевдоафинного разделения белков [14]. Схема получения препаратов рекомбинантных факторов FVIII и FIX включает несколько стадий ионообменной хроматографии, аффинную хроматографию с использованием иммобилизованных моноклональных антител, инактивацию вирусов при помощи обработки растворителем и детергентом или пастеризацией в присутствии детергента. Рассмотрим их на примере получения rFIXFc – рекомбинантного белка FIX, слитого c Fc-доменом IgG1 человека (таблица 7).
Таблица 7 – Схема получения препаратов rFIXFc [50]
|
Этап |
Назначение этапа |
|
0 |
Оттаивание флакона с клеточной линией продуцента rFIXFc |
|
1 |
Подращивание клеточной линии во флаконе |
|
2 |
Посев подращенной клеточной линии в биореактор |
|
3 |
Выращивании клеток, синтез rFIXFc |
|
4 |
Сбор клеток и клеточного дебриса центрифугированием |
|
5 |
Фильтрация супернатананта |
|
6 |
Сорбирование rFIXFc на хроматографической колонке (сapture chromatography) за счет аффинного сродства Fc-участка молекулы к носителю (Mab-Select SuReTM; GE Healthcare Bio-Sciences AB, Bj€orkgatan, Uppsala, Sweden) |
|
7 |
Элюция rFIXFc с сорбента |
|
8 |
Очистка rFIXFc от примесей балластных веществ с помощью анионообменной хроматографии |
|
9 |
Очистка rFIXFc от примесей балластных веществ с помощью псевдоафинной хроматографии |
|
10 |
Освобождение продукта от вирусов фильтрацией через фильтры с размерами пор 15 нм (Planova 15N; Asahi Kasei Bioprocesses, Inc., Glenview, IL, USA) |
|
11 |
Концентрирование продукта ультрафильтрацией или диафильтрацией |
|
12 |
Фасовка препарата rFIXFc и хранение |
Полученный рекомбинантный фактор свертывания крови валидируют для подтверждения его идентичности, чистоты, качества и активности (таблица 8).
Таблица 8 – Валидация рекомбинантных факторов свертывания крови на примере получения rFIXFc [50]
|
Тест |
Что подтверждается |
|
Полиакриламидный гель-электрофорез препарата (редуцирущий и нередуцирущий) |
Подтверждает молекулярную массу и чистоту продукта, отсутствие (наличие) агрегатов молекул |
|
Гель-хроматография препарата |
То же |
|
Коагуляционная активность (время образования и активности тромбопластина препарата, измеряется в МЕ/нмоль) |
Подтверждает специфическую активность препарата, не менее 6,5–7,1 МЕ/нмоль |
|
FcRn-связывание с rFIXFc |
Дополнительный критерий специфической активности препарата, от 104 до 124% |
|
Определение активности rFIXFc в препарате |
Уровень активности rFIXFc в пределах 0,002–0,004 мол%) |
|
Определение бионагрузки |
Безопасность препарата. Микробиологическое исследование выполняется в соответствии с руководящими принципами фармакопеи |
|
Определение эндотоксинов |
Безопасность препарата. Используется кинетический турбидиметрический метод в соответствии руководящими принципами фармакопеи |
Эффективность процесса удаления вирусов оценивают на иных моделях, чем используются при получении препаратов факторов свертывания крови из плазмы человека. Обычно используются четыре модели: ксенотропный вирус мышиной лейкемии (Х-MLV), минут-вирус мышей (ММВ), ортореовирус млекопитающих 3 (Рео-3; также известный как реовирус серотипа 3) и вирус псевдобешенства (SuHV-1). Исследования проводятся в соответствии с International Conference on Harmonisation Q5A-E Guidelines и US Food and Drug Administration Points to Consider [53–55].
***
В настоящее время нельзя считать преодоленными все проблемы, возникающие при получении препаратов медицинского назначения из плазмы крови человека и сывороток крови животных. Анализ технологий получения таких препаратов по «опережающим объектам», показывает, что отрасль и находится в постоянном поиске новых путей очистки белков плазмы крови, которые были бы более щадящими и гарантировали вирусную безопасность препаратов. В тоже время нельзя не заметить исчерпание возможностей дальнейшего развития базовых технологий очистки препаратов крови за пределы частных усовершенствований. Параллельно происходит развитие технологий, исключающих необходимость работы с донорской кровью и гипериммунными сыворотками животных, позволяющих получать препараты крови по технологиям генной инженерии. Принципиально меняются и сами препараты. На рынок выходят рекомбинантные факторы крови с измененными свойствами; коктейли из рекомбинантных антител и Fab-фрагментов IgG, высокоафинных к эпитопам токсинов и др. Происходит вытеснение этими препаратами препаратов, получаемых из плазмы человека и животных. Поэтому в ближайшие годы в России необходимо создавать принципиально новую систему оценки качества, эффективности и безопасности препаратов крови, учитывающую дальнейшее направление их развития.
ЛИТЕРАТУРА
1. Кольцова КЛ. Использование патентной документации на различных этапах научно-исследовательских и конструкторских разработок. М.; 1984.
2. Александров ЛВ, Карпова НН. Методы прогнозирования технических решений с использованием патентной информации. М.; 1991.
3. Донюш ЕК. Использование внутривенных иммуноглобулинов в клинической практике. Вопросы современной педиатрии 2011; (2): 49–63.
4. Radosevich M, Burnouf T. Intravenous immunoglobulin G: trends in production methods, quality control and quality assurance. Vox Sanguinis 2010; 98(1): 12–28.
5. Галактионов ВГ. Эволюционная иммунология. М.; 2005.
6. Борисевич ИВ, Авдеева ЖИ, Алпатова НА, Давыдов ДС, Гайдерова ЛА, Горбунов МА, с соавт. Медицинские иммунобиологические препараты. М.; 2011.
7. Oncley JL, Melin M, Richert DA, Cameron JW, Gross PM: The separation of the antibodies, isoagglutinins, prothrombin, plasminogen and beta1-lipoprotein into subfractions of human plasma. J Am Chem Soc. 1949; 71(2): 541–50.
8. Marcia C, Zago N, Carla LD. Immunoglobulin: production, mechanisms of action and formulations. Rev Bras Hematol Hemoter. 2011; 33(5): 377-82.
9. Зубкова НБ. Биотехнологические аспекты эффективной и безопасной переработки донорской плазмы: проблемы и перспективы. Биопрепараты 2014; (1): 4–10.
10. Bertolini J. Chromatographic purification of immunoglobulins downstream. Biotechnol Prog. 2000; 31: 20–1
11. Teschner W, Butterweck HA, Auer W, Muchitsch EM, Weber A, Liu SL, et al. A new liquid, intravenous immunoglobulin product (IGIV 10%) highly purified by a state-of-the-art process. Vox Sang. 2007; 92(1): 42–55
12. Terpstra FG, Parkkinen J, Tolo H, Koenderman AH, Ter Hart HG, von Bonsdorff L, et al. Viral safety of Nanogam, a new 15 nm-filtered liquid immunoglobulin product. Vox Sang. 2006; 90(1): 21–32
13. Stucki M, Boschetti N, Schafer W, Hostettler T, Kasermann F, Nowak T, et al. Investigations of prion and virus safety of a new liquid IVIG product. Biologicals 2008; 36(4): 239–47.
14. Burton SJ, Baldes B, Curling J, Haves TK, Chen D-N, Bryant C. Sequential protein isolation and purification shemes by affinity chromatography. Patent WO n 2006/023831; 2006.
15. Hooper JA: Intravenous immunoglobulins: evolution of commercial IVIG preparations. Immunol Allergy Clin North Am. 2008; 28(4): 765–78.
16. Углова ТА. Подкожное введение иммуноглобулина при заместительной терапии у детей с первичными иммунодефицитами. Медицинские новости. 2014; (5):47–51.
17. Jolles S, Stein M,∙Longhurst H,∙ Borte M,∙Ritchie B, Sturzenegger M, Berger M. New frontiers in subcutaneous immunoglobulin treatment. Biol Ther. 2011; 1(1): 003. URL: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3873072/pdf/13554_2011_Article_9.pdf
18. Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. 2007; 4(4): 427–40.
19. Yap PL: Intravenous immunoglobulin and hepatitis C virus: an overview of transmission episodes with emphasis on manufacturing data. Clin Ther. 1996; 18(Suppl B): 43–58.
20. Панов ВП. Принципы обеспечения вирусной безопасности продуктов крови. Химико-фармацевтический журнал 2004; 38(3): 39–47.
21. Tsen S, Kingsley DH., Kibler K, Sizemore S, Sara M. Vaiana P. Pathogen Reduction in Human Plasma Using an Ultrashort Pulsed Laser. PLoS/Ohe 2014 9(11): e111673.
22. Kim S, Yong WC, Yong K, Hark MS, Ki W, Yong-Sung K. Improvement of virus safety of an antihemophilc factor IX by virus filtration process. J. Microbiol. Biotechnol. 2008; 18(7): 1317–25
23. Norrby E. Prions and protein-folding diseases. J Intern Med. 2011; 270(1): 1–14.
24. Покровский ВИ, Киселев ОИ, Черкасский БЛ. Прионы и прионные болезни. М.; 2004.
25. Шкундина ИС, Тер-Аванесян МД. Прионы. Успехи биологической химии 2006; 46: 3–42.
26. Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004; 203(3): 733–9.
27. Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Dabaghian R, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013; 347: f5675.
28. Legname G, Trian Le NT, Giachin G. Synthetic prion. In. Prusiner SB. Prions and prion diseases: new developments. 2012; Nova Science Pablishers, Inc. 61–83
29. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363(9407): 417–21.
30. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364(9433): 527–9.
31. Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, et al. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 2010; 16(2): 296–304.
32. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411(6839); 810–13.
33. Lacroux C, Comoy E, Moudjou M, Perret-Liaudet A, Lugan S, Litaise C, et al. Preclinical Detection of Variant CJD and BSE Prions in Blood. PLoS Pathog 2014; 10(6): e1004202. Available from: http://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1004202 (cited 2015 July 10].
34. Surround Optical Fiber Immunoassay. Available from: https://en.wikipedia.org/wiki/Surround_Optical_Fiber_Immunoassay [cited 2015 July 10].
35. Rubenstein R, Chang B, Gray P, Piltch M, Bulgin MS, Sorensen-Melson S, Miller MW. Prion Disease Detection, PMCA Kinetics, and IgG in Urine from Sheep Naturally/Experimentally Infected with Scrapie and Deer with Preclinical/Clinical Chronic Wasting Disease. J Virol. 2011; 85(17): 9031–8.
36. Prusiner SB. Prion protein standard end method of making same. Патент Канады, № 2349337; 2000.
37. Schwartz R. Overview of the biochemistry and safety of a new native intravenous gammaglobulin, IGIV, pH 4,25. Amer J Med. 1987; 83 (4A): 46–52.
38. Абрамова ЕГ, Никифоров АК, Лобовикова ОА, Еремин СА, Васин ЮГ, Михеева ТА, с соавт. Производство гетерологичного антирабического иммуноглобулина – итоги первых пяти лет. Проблемы особо опасных инфекций 2010; 3(105): 58–62.
39. Ситник НП. Разработка высокоочищенного препарата иммуноглобулина антирабического из плазмы крови лошади дис. ... канд биол. наук. 2007 Available from: http://www.dissercat.com/content/razrabotka-vysokoochishchennogo-preparata-immunoglobulina-antirabicheskogo-iz-plazmy-krovi-l.
40. Alvarenga LM, Zahid M, di Tommaso A, Juste MO, Aubrey N, Billiald P, Muzard J. Engineering Venom's Toxin-Neutralizing Antibody Fragments and Its Therapeutic Potential. Toxins. 2014; 6(8): 2541–67.
41. Goyffon M. Passive immunotherapy today: Brief history. Biol Aujourdhui. 2010; 204(1): 51–4.
42. Орлов БН, Гелашвили ДБ. Зоотоксинология (ядовитые животные и их яды). М.; 1985.
43. Li D, Mattoo P, Keller JE. New equine antitoxins to botulinum neurotoxins serotypes A and B. Biologicals 2012; 40(4): 240–6.
44. Werdan K, Pilz G, Muller-Werdan U, Enriquez MM, Schmitt DV, Mohr FW, et al. Immune globulin G treatment of post cardiac surgery patients with score-identified severe SIRS — The ESSICS study. Crit Care Med. 2008; 36(3): 716–23.
45. Lou J, Geren I, Garcia-Rodriguez C, Forsyth CM, Wen W, Knopp K, et al. Affinity maturation of human botulinum neurotoxin antibodies by light chain shuffling via yeast mating. Protein Engineering, Design & Selection 2010; 23(4): 311–19.
46. Burnouf T. Recombinant plasma proteins. Vox Sanguinis 2011; 100(1): 68–83.
47. Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt B, et al. Expression of active human factor VIII from recombinant DNA clones. Nature 1984; 312(5992): 330–7.
48. Kurachi K, Davie EW: Isolation and characterization of a cDNA coding for human factor IX. Proc Natl Acad Sci USA. 1982; 79(21): 6461–4.
49. Орлова НА, Ковнир СВ, Воробьев ИИ, Габибов AГ, Воробьев АИ. Фактор свертывания крови VIII – от эволюции к терапии. Acta naturae 2013; 5(2): 19–39.
50. Mccue J, Osborne D, Dumont J, Peters R, Mei B, Pierce GF et al. Validation of the manufacturing process used to produce long-acting recombinant factor IX Fc fusion protein. Haemophilia 2014; 20(4): 327–35.
51. Peyvandi F, Garagiola I, Seregni S. Future of coagulation factor replacement therapy. Journal of Thrombosis and Haemostasis 2013; 11(Suppl. 1): 84–98.
52. Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood 2010; 115(10): 2057–64.
53. Food and Drug Administration. CFR- Code of Federal Regulations Title 21, Part 58-Good Laboratory Practice. Available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=58. Accessed september 3, 2013.
54. International Conference on Harmonisation (ICH). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. Q5A(R1). Available at http://private.ich.org/LOB/media/MEDIA425.pdf. Accessed september 3, 2013.
55. Department of Health and Human Services (DHHS). Center for Biologics Evaluation and Research. Points to consider in the characterization of cell lines used to produce biologicals. Available at http://www.fda.gov/downloads/biologicsbloodvaccines/safetyavailability/ucm162863.pdf. Accessed september 3, 2013.
Библиографическое описание: Супотницкий МВ, Елапов АА, Борисевич ИВ, Кудашева ЭЮ, Климов ВИ, Лебединская ЕВ, Корсун ЛВ, Горбунова ЕВ, Слободян ВГ, Меркулов ВА, Олефир ЮВ. Препараты, полученные из крови человека и животных, в аспекте показателей качества, эффективности и безопасности. Биопрепараты 2015; (3): 33–48.
Bibliographic description: Supotnitskiy MV, Elapov AA, Borisevich IV, Kudasheva EYu, Klimov VI, LebedinskayaEV, Korsun LV, Gorbunova EV, Slobodyan VG, Merkulov VA, Olefir YuV. Blood preparations of humans and animals in terms of their quality, efficacy and safety. Biopreparation (Biopharmaceuticals) 2015; (3): 33–48.
Ссылки по теме:
[1] Гликозилирование — ферментативный специфический процесс, в ходе которого происходит присоединение остатков сахаров к органическим молекулам. Гликозилирование является одной из форм котрансляционной и посттрансляционной модификации белков. Преобладающая часть белков, синтезируемых в шероховатом эндоплазматическом ретикулуме, подвергается гликозилированию.

Михаил Васильевич Супотницкий
Российский микробиолог, полковник медицинской службы запаса, изобретатель, автор книг и статей по истории эпидемий чумы и других особо опасных инфекций, истории разработки и применения химического и биологического оружия. Заместитель главного редактора научно-практического журнала «Вестник войск РХБ защиты» Министерства обороны РФ.